Toll-like receptor 3 signalling mediates angiogenic response upon shock wave treatment of ischaemic muscle
Aims
Shock wave therapy (SWT) represents a clinically widely used angiogenic and thus regenerative approach for the treatment of ischaemic heart or lim disease. Despite promising results in preclinical and clinical trials, the exact mechanism of action remains unknown. Toll-like receptor 3, which is part of the innate immunity, is activated by binding doublestranded (ds) RNA. It plays a key role in inflammation, a process that is needed also for angiogenesis. We hypothesize that SWT causes cellular cavitation without damaging the target cells, thus liberating cytoplasmic RNA that in turn activates TLR3.
Methods and results
SWT induces TLR3 and IFN-b1 gene expression as well as RNA liberation from endothelial cells in a time-dependant
manner. Conditioned medium from SWT-treated HUVECs induced TLR3 signalling in reporter cells. The response was
lost when the medium was treated with RNase III to abolish dsRNAs or when TLR3 was silenced using siRNAs. In a
mouse hind limb ischaemia model using wt and TLR32/2 mice (n ¼ 6), SWT induced angiogenesis and arteriogenesis
only in wt animals. These effects were accompanied by improved blood perfusion of treated limbs. Analysis of main
molecules of the TLR3 pathways confirmed TLR3 signalling in vivo following SWT.
Conclusion
Our data reveal a central role of the innate immune system, namely Toll-like receptor 3, to mediate angiogenesis upon release of cytoplasmic RNAs by mechanotransduction of SWT.
———————————————————————————————————————————————————–
Keywords
Toll-like receptor 3 † Angiogenesis † Shock wave therapy
- Introduction
Ischaemic heart and limb disease are prevalent clinical problems in industrialized countries. Both represent an increasing global socioeconomic health burden.1 – 3 The morphological surrogate of chronic
ischaemia, caused by atherosclerosis of coronary or peripheral arteries, is mainly due to loss of myocytes leading to functional deterioration.
Endogenous neovascularization following ischaemia is often insufficient. Regenerative therapies aim to safe hibernating myocytes at ischaemic border zones by induction of angiogenesis. Current strategies mainly include gene and (stem) cell-based approaches.4,5
Both showed promising results in animal studies and partly in clinical application. However, none of them yet gained broad clinical use. Thus, effective strategies that are clinically easy to implement and
that have a favourable side effect profile are of high interest. A new approach to induce angiogenesis in ischaemic tissue is lowenergy shock wave therapy (SWT).6 – 8 SWs represent a specific type
of sound pressure waves being used in medicine for over 30 years for lithotripsy. Due to this long-term clinical application, its safety profile is quite well studied, and hence, SWT is well proven not to cause any severe side effects or even neoplastic tissue transformation. Moreover, SWT has been shown to be effective in chronic and acute myocardial ischaemia where it was found to reduce angina symptoms and improve left ventricular ejection fraction in patients suffering from coronary artery disease.9,10 It has been shown to enhance recruitment and homing of bone marrow-derived mononuclear cells in preclinical and clinical trials.11,12. Besides knowledge about induction of angiogenesis and functional restoration of the target tissue, the mechanism of how the mechanical
stimulus of SWs is translated into a biological response remains largely unknown. SWs have earlier been described to cause an increase of cell
membrane permeability.13,14 Therefore, we suggested that this might trigger the release of cytoplasmic RNA and that this released doublestranded (ds) RNA would stimulate Toll-like receptor 3 (TLR3) in healthy adjacent cells. TLR3 is a pattern recognition receptor that has first been described to detect double-stranded viral RNAs. More recently, it was shown to act as a ligand for mRNA, microRNAs, and siRNAs.15 – 17 It is part of the innate immune system and modulates inflammation by an array of inflammatory cytokines and Type 1 IFNs.
Its signalling pathway also results in the activation of the transcription factor nuclear factor-kappa B (NF-kB) which itself induces transcription of IFN-b1.18 TLR3 was found to be expressed on endosomes and on the surface of endothelial cells.19,20 The induction of angiogenesis is depending on an initial inflammatory
process that is characterized by the invasion of a broad variety of cell types like macrophages, neutrophils, fibroblasts, and endothelial progenitor cells.21,22 Hence, it seems to be reasonable that evolutionary highly conserved systems like the innate immune system may play a pivotal role as driving forces for angiogenesis.
Methods
2.1 Animals
Experiments were approved by the Austrian animal care and use committee. The investigation conformed to the ‘Guide for the Care and Use of Laboratory Animals’ published by the US National Institutes of Health
(NIH Publication No. 85-23, 1996, revised 2011; available from: www.nap.edu/catalog/5140.html). The investigators performed measurements and analyses in a blinded fashion. Twelve- to 14-week-old male C57/BL6 wild type (Charles River, Sulzfeld, Germany) and TLR3 knockout mice (TLR32/2, C57/BL6 background) mice
weighing 25 – 30 g were randomly assigned to the treatment groups. The sample size of each group at each measurement point was n ¼ 6. During
the experiments, mice were housed under standard conditions with a 12-h light/dark cycle. Water and commercial mouse diet were available ad libitum.
2.2 Hind limb ischaemia model and perfusion
measurement
Anaesthesia was administered by an intraperitoneal injection of ketamine
(80 mg/kg body weight) and xylazine hydrochloride (5 mg/kg body weight).
Femoral artery was ligated and excised between the inguinal ligament and
proximal to the branching into saphenous and popliteal artery using 7 – 0 polypropylene sutures (Ethicon, USA). Perioperative mice received piritramide (10 mg/kg body weight) and tramadolhydrochloride (2.5 mg/100 mL drinking water) for the first 7 days after operation. The physical condition of the animals was evaluated two times per day. Blood flow measurements were performed prior to surgery and each
week thereafter using a laser Doppler perfusion image analyser (Moor Instruments, USA) as previously reported.23 To minimize data variables attributable to temperature, mice were kept on a heating plate at 378C
before measurement. Blood perfusion was expressed as the ratio of left (operated, ischaemic leg) to right (not operated, non-ischaemic leg) limb blood flow. A ratio of 1 prior to surgery indicated equal blood perfusion in both legs. Limb necrosis score was determined as described previously.24 Briefly, mice were investigated 4 weeks post induction of hind limb ischaemia and scored with 0 points if no necrosis or defect was observed, with 1 point if skin necrosis was present, with 2 points if below ankle amputation was present, and with 3 points if above ankle amputation was observed.
Animals were sacrificed 72 h, respectively, 28 days after SWT via cervical dislocation. Muscle samples were obtained as described previously.6 Subsequently, they were rinsed in sterile saline and divided into equal halves, one part of which was placed in 4% paraformaldehyde (PFA) overnight, and the other part was snap frozen in liquid nitrogen.
2.3 SW treatment
Immediately after hind limb ischaemia surgery, a single SWT was performed in the treatment group, whereas control animals were kept untreated. The commercially available Orthogold device with applicator CG050-P (Tissue Regeneration Technologies Llc., USA) was used for SWT. Three hundred impulses at an energy flux density of 0.1 mJ/mm2 and a frequency of 4 Hz were applied to skin at the area of the adductor muscle. Common ultrasound gel was used for coupling of the SW applicator. Cells were treated in a specifically designed water bath, which guaranteed avoidance of disturbing reflections and constant temperature at 378C. Cell culture flasks were filled with culture medium prior to treatment, as SWs would be reflected by air. Culture flasks were subsequently dunked into the water bath and treated at 7 cm distance to the applicator. Thus, 300 impulses at an energy flux density of 0.08 mJ/mm2 and a frequency of 3 Hz were applied. According to the applied number of impulses and the used frequency, the raw time of SW application in vivo is 1 min and 40 s (3 impulses per second, 300 impulses in toto). Including the exhibition of the region of interest per animal, hair removal, and gel application, the whole procedure takes in average 3 min per animal.
2.4 Histopathologic examination
Paraformaldehyde-fixed (4%) and paraffin-embedded femoral muscles were sectioned at 5 mm thickness. Slides were stained with Mayer’s haematoxylin according to standard procedures.25 Briefly, slides were deparaffinized in xylene (Merck, Germany) and subsequently rehydrated in decreasing alcohol concentrations. After rehydration, H&E was incubated
for 5 min on the slides. To develop the characteristic blue nuclei staining, the slides were incubated in tap water for 10 min. After subsequent dehydration in increasing alcohol concentrations, slides were mounted in DPX (Merck, Germany). Histological examinations were conducted in a blinded fashion; for each slide, three fields at ×400 magnification were photographed from the region at risk. Images were taken with the Leica DM5000B microscope.
2.5 Cell culture
After obtaining written informed consent of patients, umbilical cords were obtained from Caesarean sections for isolation of human umbilical vein endothelial cells (HUVECs). Permission was given from the ethics committee of Innsbruck Medical University (no. UN4435) and complied to the Declaration of Helsinki. Isolation of endothelial cells was performed
as described in detail previously.26 Briefly, cords were mechanically meshed using sterile scissors. The mesh was then passed through a sieve to eliminate cell lumps or conjunctive tissue rests. HUVEC monocultures were
obtained by separating the ECs using magnetic beads that specifically bind endothelial cells (MACS, Germany). The quality of the obtained monoculture was verified using FACS analyses on CD31 (BD, Germany). Freshly isolated HUVECs were then cultivated in endothelial cell basal medium (CC-3156, Lonza, Switzerland) supplemented with EGM-2. SingleQuots supplements (CC-4176, Lonza, Switzerland) at a temperature of 378 with 0.5% CO2 and a saturated air humidity. Prior to SWT treatment, cells were starved in serum-free medium for 24 h. After SWT, cells were
harvested 2, 4, and 6 h and processed for further analyses. Two hundred microgram per millilitre of the structural analogue to double-stranded RNA polyinosinic : polycytidylic acid [Poly (I : C)] (InvivoGen, San Diego, CA, USA) served as a positive control for TLR3 activation in HUVECs. For ds RNA depletion, the medium from HUVECs treated with SWT
was incubated with 4 U/mL of RNase III (Biozym, Germany) for 1 h at 378C in a cell culture incubator under standard conditions. The conditioned medium was then heated up to 508C for 10 min to deactivate the RNase III prior to incubation on the reporter cells. For inhibition of protein biosynthesis, HUVECs were incubated with 10 mg/mL cycloheximide (CHX)
(Sigma-Aldrich, USA) for 30 min prior to SWT. Cells were harvested 6 h after SW treatment.
RNA was isolated using the RNeasy kit (QIAGEN, Germany) from treated and untreated cells supernatant and put on HUVECs for another 24 h to prove that RNA is causing TLR3 stimulation. For RNA –protein complex analysis, RNA was added with either 4 U/mL RNase III alone or RNase III plus 25 mg/mL Proteinase K (Sigma, USA).
2.6 Small-interfering RNA
HUVECs were transiently transfected with small interfering RNA (siRNA) for TLR3 (no. SI02655156 SI) or AllStars Negative Control siRNA (scrambled siRNA) (both QIAGEN, Germany) according to the manufacturer’s protocol. Transfection was performed using HiPerFect (QIAGEN, Germany) for 3 h. Efficacy of knockdown was assessed 48 h after transfection by RT –PCR. Hs_TLR3_8_HP validated siRNA sequence:
AAGAACTGGATATCTTTGCCA.
2.7 Luminometer assay
A stable HEK (human embryonic kidney) reporter cell line (TLR3/ISRELUCPorter; Imgenex, USA) was purchased for luminometer assay experiments. Cells were cultivated according to the manufacturer’s protocol in DMEM with 4.5 g/L glucose (Lonza, Switzerland), 10% FCS (Sigma, USA), penicillin/streptomycin, and 4 mM L-glutamine and 3 mg/mL puromycin (Gibco, USA) as selection agent. Cells were treated with SWT as described above or incubated with supernatant from treated HUVECs (supernatant obtained 24 h after SWT). Two hundred micrograms per millilitre of TLR3 agonist Polyinosinic : polycytidylic acid [Poly(I:C)] (Invivogen, USA) were used as positive control. Twenty-four hours after treatment, cells were washed with PBS, and luminescence was analysed using a Luciferase Assay System (Promega, USA). For the detection of luminescence, Hidexw Chameleon Microplate Reader(Hidex, Finland) was used.
2.8 LDH assay
To examine cell damage, the amount of lactate dehydrogenase (LDH) release of treated cells was quantified as described previously.27 Briefly, the LDH cytotoxicity kit II (Biovision, USA) was used. LDH is indirectly measured by an enzymatic coupling reaction. LDH oxidizes lactate to generate NADH, which then generates a yellow colour. The intensity of the generated colour correlates directly with the cell number lysed. The intensity of the colorimetric reaction is then quantified using a Hidexw Chameleon Microplate Reader (Hidex, Finland) plate reader at OD 450 nm. Sham-treated cells served as a control. Supernatants were analysed immediately after SWT, 30 min, 6 h and 24 h after SWT. All measurements were performed in triplicate.
2.9 Immunofluorescence
Five micrometre tissue paraffin sections were deparaffinized. Subsequently, heat-mediated antigen retrieval using unmasking solution (VECTORw antigen unmasking solution, VECTOR Laboratories, USA) was performed according to the manufacturer’s protocol. Samples were blocked with a mixture of serum from the host organism of the corresponding secondary antibody used, and solved in 1× TBS supplemented with 5% BSA and 0.1% Tween-20 as blocking buffer for 30 min at room temperature. For specific alpha-smooth muscle actin (a-SMA, rabbit anti-mouse, Epitomics, USA) and CD31 (rat anti-mouse SZ31, Dianova, Germany) staining, slides were incubated with the primary antibody overnight at 48C. For signal detection, secondary antibodies (donkey anti-rat IgG Alexa 488, Jackson Immuno Research, USA; goat anti-rat IgG Alexa Fluor 350, MoBiTec, Germany; donkey anti-rabbit Fab’2 PE, Fitzgerald, USA) were incubated for 1 h at room temperature. Nuclear staining was performed using DAPI containing mounting medium (Life Technologies, USA) according to the manufacturer’s protocol. Analysis of the slides was performed in a blinded fashion. For each slide,
10 randomly chosen fields at ×400 magnification were photographed. For quantification, images were taken with the Leica DM5000B microscope and analysed using an automatized MatLab algorithm (The Mathworks, USA).
The software determines the number of pixel in each RYB channel, thus measuring the relative area occupied by blue (cores) and green or red staining per image. Quantification was done by setting blue coloured pixels in proportion to the total number of otherwise coloured pixels as described elsewhere. 25 HUVECs treated with 200 mg/mL Poly (I:C) for 6 h were fixed with 4% PFA and subsequently permeabilized with 0.1% Triton (Sigma, USA) in PBS. Blocking was performed using 1% BSA in PBS for 1 h at room temperature.
Samples were incubated for 1 h at room temperature with anti-TLR3 antibody (Sigma-Aldrich, USA). Goat anti-rabbit IgG Alexa 568 antibody (Life Technologies, USA) was used for signal detection. DAPI staining and aqueous mounting (Life Technologies, USA) was performed according to the manufacturer’s protocol.
2.10 Real-time RT–PCR
Total RNA was isolated from homogenized muscle samples using TRI Reagent (Sigma-Aldrich, USA) according to the manufacturer’s protocol. Then cDNA was synthesized using iScript cDNA Synthesis kit (Bio-Rad laboratories, USA). Gene expression profiles of mouse-specific mRNA profiles (primer sequences are listed in Supplementary material online, Table
S1) were assessed by quantitative real-time PCR using a StepOne Plus realtime PCR device (Applied Biosystems, USA). Briefly, after a denaturation phase of 10 s at 958C, followed by an annealing at 608 for 10 s, a synthesis
step at 728 for 20 s was performed. Fluorescence was detected at 788C at the end of every cycle. Specificity of the primers used was detected by adding a melting curve procedure to the amplification process. For this purpose, the temperature was brought down to 608C and rose by steps of 0.58C until 958C. After every step, fluorescence was detected. Evaluation
of the specificity of the primers was performed as described elsewhere.28
2.11 Tissue, cell culture supernatant, and serum
RNA measurement
Total RNA contents from isolated tissue samples, cell culture supernatants, or from serum samples from the in vivo experiments were detected photometrically using a NanoPhotometer (Implen, Germany) device. From each
sample, 3 mL of the raw substance (medium, serum, sample template) was used to detect optical density on 230, 260, 280, and 320 nm, respectively. Each sample was measured 3 times and the mean was calculated as final concentration. The ratio A 260/280 and A260/230 was used to determine the quality of the isolated RNAs. Values above 2 were considered as high quality.
2.12 Protein isolation and western blot analysis
Proteins from frozen muscles were isolated as described elsewhere.28 Gel electrophoresis and western blotting were performed according to standard procedures. Briefly, 10 and 7.5% SDS gels were loaded with 50 mg protein per lane. The first lane was always reserved for the Spectra broad range marker (Fermentas, Germany) as standard. Proteins were then immobilized on Hybond C supermembrane (Amersham Pharmacia Biotech, UK) using the semi dry blotting technique (Bio-Rad, Germany) for 60 min at 10v. The membranes were then quality checked using Ponceau dye. After blocking of the membranes using 5 – 8% BSA in TBS substituted with 0.1% Tween-20, the blots were probed with antibodies as follows:
rabbit anti-mouse Phospho-IRAK4 (Cell Signaling Technology, USA), rabbit anti-mouse IRF3 (Thermo Fisher Scientific, Germany), rabbit anti-mouse TRIF (Thermo Fisher Scientific, Germany), rabbit anti-mouse TLR-3 (Sigma-Aldrich, USA), rabbit anti-mouse VEGF (Bio-Rad laboratories, USA). Detection was then performed by incubating the membranes with a secondary anti-rabbit HRP-labelled antibody (Vector, Germany). Chemoluminescence was induced using the Western Blot Luminol reagent (Santa
Cruz Biotechnologies, USA) and the digitalization and evaluation of the blots were performed with a Kodak Imager (Carestream, Germany).
2.13 Statistical analysis
All results are expressed as mean+SEM. Statistical comparisons between two groups were performed by Student’s t or Mann –Whitney test as appropriate. Multiple groups were analysed by two-way ANOVA followed by Bonferroni’s multiple comparison test to determine statistical significance. Probability values ,0.05 were considered statistically significant. All experiments were repeated at least in triplicate.
3. Results
3.1 SW treatment of reporter cells results
in IRF3-dependent TLR3 activation
To evaluate whether TLR3 activation is mediated by liberated cellular RNA, we incubated human embryonic kidney (HEK) TLR3 reporter cells with the supernatant of SW-treated HUVECs and with the agonist Poly(I:C) as a positive control and treated HEK cells directly with SW.
Luminescence of directly treated (arbitrary units SWT 86.45+1.07, P , 0.001 vs. CTR), supernatant incubated (89.4 +1.19, P , 0.001 vs. CTR), and Poly(I:C) incubated (71.75+3.64, P , 0.01 vs. CTR) reporter cells was significantly increased compared with untreated controls (Figure 1A). TLR3 stimulation at protein level was confirmed by western blot analysis and showed a time-dependent result with an elevation starting at 30 min after SWT increasing up to 48 h (rel. protein expression: CTR 133.51, SWT 30′ 148.59, SWT 1 h 159.06, SWT 2 h 175.09, SWT 6 h 182.85, SWT 24 h 217.36, SWT 48 h 229.20). In parallel, a time-dependent increase of total RNA content in supernatant of treated cells could be observed with a peak at 6 h after treatment (ng/mL: CTR 26.40+2.71, SWT 30′ 70.07 +15.17, SWT 1 h 87.80 + 15.33, SWT 2 h 102.25 + 9.35, SWT 6 h 135.38 + 21.62,
SWT 24 h 123.57 + 5.45, SWT 48 h 77.65 + 1.30; n ¼ 3)
(Figure 1A). To further prove whether TLR3 stimulation upon SWT is indeed mediated by RNA, we isolated RNA from untreated and treated HUVECs supernatant and incubated another HUVECs with it for 24 h. However, no significant difference in TLR3 protein could be observed (rel. protein expression: HUVEC + CTR RNA 175.40 + 3.33 vs. HUVEC + SWT RNA 191.40+0.08) (Figure 1C). As naked RNA is unstable, we therefore hypothesized that RNA might be bound in protein complexes. Another experiment with HEK reporter cells showed that RNase alone did not abolish effects, but RNase plus Proteinase K abolished TLR3 signalling upon SWT (CTR 4288 + 299.67, SWT 156603.50 + 16004.65, SWT + RNase 176046.60 + 20984.66, SWT + RNase + Proteinase K 3715.50+1545.96). This clearly indicates that released RNA forms complexes with proteins that however need to be identified in future experiments.
3.2 TLR3 gene silencing results in loss of SW response
To show that TLR3 is essential for induction of biological response upon SWT, we performed a gene knockdown in HUVECs using siRNA. TLR3 knockdown resulted in loss of response for TLR3 mRNA 6 h after treatment (arbitrary units CTR 100 + 0.99, SWT 378.3 + 11.62, scr siRNA 26.05 + 3.44, siRNA-TLR3 103.59 + 10.90, siRNA-TLR3 + SWT 106.95 +10.9; SWT vs. all other groups P , 0.001) (Figure 2A). IFN-b1 mRNA expression served as a readout analysis of downstream TLR3 activation. IFN-b1 showed a strong up-regulation in SW-treated HUVECs but no response in TLR3 silenced cells (arbitrary units CTR 19.38+0.36, SWT 131.76+5.9, scr siRNA 18.93+10.38, siRNA-TLR3 26.34+0.92, siRNA-TLR3 + SWT 9.03+1.34; SWT vs. all other groups P , 0.001) indicating that TLR3 activation is mandatory for the translation of SWT response in HUVECs (Figure 2A). The use of scrambled siRNA confirmed that
transfection itself did not alter TLR3 or IFN-b1 expression.
3.3 SW treatment of endothelial cells causes RNA release into supernatant without damaging cells
To test the hypothesis whether SWT causes a release of RNA from treated cells, we measured the total RNA content in supernatant of SW-treated HUVECs. Significantly higher amounts of RNA were found in the supernatant of SW-treated cells indicating that TLR3 activation might be triggered via RNA release into the extracellular space (in mg/mL, CTR 19.73 + 1.04 vs. SWT 34.93 + 0.87, P , 0.001; siRNA-TLR3 23.67 + 1.18 vs. siRNA-TLR3 + SWT 44.0 + 0.79, P , 0.001; scr siRNA 22.47+1.87) (Figure 2B). Notably, RNA release was as well present in TLR3-silenced but SW-treated cells indicating that RNA release caused by SWT itself is not TLR3 dependent. As cell damage could be one reason for cytosolic RNA release, we further measured LDH from treated cells and quantified it by a LDH cytotoxicity kit. Our results clearly confirm that SW-treated cells were not damaged (see Supplementary material online).
3.4 RNA but not proteins in supernatant contribute to TLR3 activation after SWT
To verify whether the effect of supernatant from SW-treated endothelial cells is indeed mediated by cellular RNA, we incubated the supernatant of SW-treated HUVECs with RNase and treated HEK TLR3 reporter cells therewith. The effect on TLR3 activation was almost completely abolished (luminescence in arbitrary units: CTR 10.17+
2.91, CTR + RNase 8.71+1.02, SWT 29.47+7.43, SWT + RNase 15.37 + 2.88, Poly(I:C) 22.02 + 4.94, Poly(I:C) + RNase 11.27 + 3.61). These data indicate that RNA is responsible for TLR3 activation (Figure 2C).
Moreover, TLR3 stimulation after SWT was not impaired in
CHX-incubated HUVECs, neither was this the case for IFN-b1 and for angiogenic factors VEGF and Tie-2 mRNA up-regulation (see Supplementary material online). These results confirm that proteins are not involved in SW-mediated TLR3 stimulation.
3.5 Angiogenic response to SWT is missing
in TLR3 knockout mice
Having found the high impact of SWT on TLR3 in vitro, we aimed to verify these findings in vivo. Therefore, we induced hind limb ischaemia
(HLI) in wild-type and TLR3 knockout mice (TLR32/2). SWs were applied immediately after induction of hind limb ischaemia to the ischaemic limb in the treatment group (SWT). Ischaemic limbs of untreated animals served as a control (CTR). Same groups were performed in TLR32/2 mice: TLR32/2 treatment group (TLR32/2 + SWT) and untreated TLR32/2 mice (TLR32/2). As known from previous experiments angiogenesis following SWT is mainly mediated by VEGF. Therefore, we first analysed VEGF receptors 1 and 2 mRNA in ischaemic muscle extracts. VEGFR 1 mRNA levels were not significantly increased (CTR 55.8+18.49, SWT 36.05 + 7.04, TLR32/2 107.0 + 8.95, TLR32/2 + SWT 106.33 + 10.83) (Figure 3A). In contrast, VEGFR2 mRNA expression was increased significantly in SW-treated wild-type animals compared with TLR32/2 mice (CTR 24.18 +4.53, SWT 120.19 + 20.25, TLR32/2 18.99 + 4.69, TLR32/2 + SWT 19.56+3.9, P , 0.001) (Figure 3B). Higher levels of VEGF-A protein could be found in the wild-type SW-treated group compared with untreated controls and TLR32/2 animals (Protein expression determined in arbitrary units; CTR 16.76+0.06 vs. SWT 63.06+8.23, P , 0.01; TLR32/2 33.1+1.3, TLR32/2 + SWT 37.07+0.57, both P , 0.05 vs. SWT) (Figure 3C). Placental growth factor (PlGF) is necessary to recruit smooth muscle cells for maturation of vessels by means of arteriogenesis. PlGF mRNA was highly upregulated in SW treated wild types compared with all other groups (CTR 126.45+17.42 vs. SWT 374.67+126.25, P , 0.01; TLR32/2 118.46 + 24.07 vs. TLR32/2 + SWT 108.38 + 12.39, P , 0.01)
(Figure 3D). The endothelial cell-specific receptor Tie-2 mRNA was highly increased indicating involvement of the angiopoietin/Tie-2 system in SWT-induced angiogenesis (arbitrary units, CTR 2037.2 + 202.32 vs. SWT 69860.0 +2849.28 and TLR32/2 42.18+ 8.52 vs. TLR32/2 + SWT 74.11+26.66, both P , 0.001) (Figure 3E).
3.6 Functional recovery and induction of angiogenesis after SWT is deteriorated in TLR3 knockout mice
To assess functional recovery, we measured limb perfusion before and after hind limb operation and weekly thereafter for 4 weeks using Laser Doppler imaging. Perfusion is expressed as ratio of ischaemic vs. non-ischaemic limb. SW-treated TLR3 knockout mice showed no significant improvement of perfusion ratio after hind limb ischaemia compared with untreated controls (3 weeks following SWT: TLR32/2 0.59+0.04 vs. TLR32/2 + SWT 0.54+0.08, P . 0.05; 4 weeks following SWT: TLR32/2 0.53 + 0.02 vs. TLR32/2 + SWT 0.52 +0.07, P . 0.05), whereas SW-treated wild-type animals improved significantly 3 and 4 weeks after treatment (3 weeks: CTR 0.51+0.06vs. SWT 0.82+0.09, P , 0.05; 4 weeks: CTR 0.48+0.08 vs. SWT 0.78+0.03, P , 0.01) (Figure 4A and B). Notably, untreated wild-type controls recovered better than TLR32/2 mice did after 1 and 2 weeks. Necrosis score (amount of peripheral limb necrosis) was assessed 4
weeks after hind limb ischaemia induction. SW-treated wild-type mice showed a significant decrease in necrosis score compared with their untreated controls and TLR32/2 mice (CTR 1.0 + 0.26 vs. SWT 0.29 + 0.2, P , 0.05). No improvement of necrosis was found in TLR32/2 mice, neither with SWT (TLR32/2 1.6 + 0.22 vs. TLR32/2 + SWT 1.67+0.21, P . 0.05) (Figure 4C). In line with this, CD31 and smooth muscle cells (a-SMA) were both
highly expressed in SW-treated wild-type mice at 4 weeks after treatment. The effect of increased angiogenesis and arteriogenesis by SWT was abolished in TLR32/2 mice (Figure 5A). CD31-positive endothelial cells (CTR 0.41 + 0.15, SWT 2.94 + 1.67, TLR32/2 0.23 + 0.04, TLR32/2 + SWT 0.42+0.14, P , 0.05) (Figure 5B). a-SMA-positive smooth muscle cells: (CTR 4.91+1.06, SWT 13.63+4.0, TLR32/2 0.42+0.14, TLR32/2 + SWT 0.46+0.06, P , 0.05) (Figure 5C).
3.7 SW treatment stimulates TLR3 signalling in vivo
To prove TLR3 stimulation in vivo, we measured TLR3 and IFN-b1 mRNA in SW-treated and untreated wild-type mice. Seventy-two hours after induction of hind limb ischaemia and SW treatment TLR3 mRNA was significantly increased in SW-treated animals compared with untreated controls (CTR 17.62+3.35 vs. SWT 116.67+36.5, P , 0.001) (Figure 6A). To show TLR3 downstream activation, we studied IFN-b1 expression as being a well-known TLR3 readout gene.
IFN-b1 mRNA expression was significantly increased 72 h after induction of hind limb ischaemia and SW treatment (mRNA levels expressed in arbitrary units; CTR 156.35+ 28.8 vs. SWT 1067.35 + 123.67, P , 0.001). No increase in IFN-b1 mRNA levels could be observed in any of the TLR32/2 groups (TLR32/2 26.48 + 11.26, TLR32/2 + SWT 20.56 + 3.55) (Figure 6A). As circulating RNA is meant to be responsible for TLR3 stimulation upon SWT, we measured its levels in the serum and found a significant increase in SW-treated
animals (RNA in mg/mL: CTR 496.30 + 14.97 vs. SWT 583.67 + 3.78, P , 0.001; TLR32/2 341+21.81 vs. TLR32/2 + SWT 417+ 23.74, P , 0.05) (Figure 6B). Serum then was put on cultured HUVECs to verify whether circulating RNAs are capable of activating TLR3. Indeed, we found a significant up-regulation of TLR3 mRNA (arbitrary units: CTR 306.53 + 70.35, SWT 940.04 + 305.77, CTR + RNase 240.95 + 26.56, SWT + RNase 283.93 + 49.89; all P , 0.01 vs. SWT) as well as of downstream gene IFN-b1 (arbitrary units: CTR 28.91 + 11.76, SWT 466.68 + 179.32, CTR + RNase 152.23 + 15.33, SWT + RNase 156.40+0.14; all P , 0.01 vs. SWT). This effect again was abolished by RNase (Figure 5B).
Higher amounts of TLR3 could be seen in IF staining of ischaemic muscle at 72 h after SW treatment compared with hardly any TLR3 in untreated controls (% positive cells: CTR 3.48 + 0.69 vs. SWT 25.70+4.02, P ¼ 0.029) (Figure 6C).
Transcription factor NF-kB, that induces transcription of IFN b1, gets activated by a TRIF (TIR-domain-containing adapter-inducing IFN-b)-dependent pathway, which is utilized by TLR3 and TLR4.18 A simplified TLR3 and TLR4 pathway scheme is shown in Figure 6D. Therefore, we first aimed to measure TRIF protein expression in muscle tissue 72 h after treatment. Relative protein expression of TRIF was
significantly increased in SW-treated wild types compared with TLR32/2 mice with and without SWT (CTR 33.78 +8.54 vs. SWT 88.47+7.62, P , 0.05; TLR32/2 33.11+8.98 vs. TLR32/2 + SWT 24.91 + 3.12, P . 0.05; SWT vs. TLR32/2 + SWT, P , 0.001) (Figure 6E). TRIF recruits and directly binds TRAF6 (TNF receptor-associated factor 6), which was found to be significantly up-regulated after 72 h at mRNA level in SW-treated wild-type mice compared with TLR32/2 mice with and without SWT (CTR 112.38 + 19.29 vs. SWT 483.7 + 121.96, P , 0.001; TLR32/2 70.61 + 11.38 vs. TLR32/2 + SWT 53.99+3.4, P . 0.05; SWT vs. TLR32/2 + SWT, P , 0.001) (Figure 6F). TRAF6 is addressed in both TLR3 and TLR4 signalling. However, in the TLR4 pathway, it is dependent on IRAK4 (IL-1 receptor-associated kinase 4), as it gets recruited by MyD88 (myeloid differentiation primary response gene) via the protein kinase IRAK4. To exclude the involvement of TLR4 signalling after SW treatment, we measured IRAK4
mRNA and as well as its activated protein, phosphoIRAK4. Both showed no difference between treatment or control groups in wildtype mice, clearly indicating that TLR4 signalling is not involved after SW treatment. However, in TLR32/2 mice, TLR4 signalling could be found indicating that TLR4 serves as a rescue pathway if TLR3 signalling
is impaired (see Supplementary material online). In TLR3 signalling, TRIF not only recruits TRAF6 but also recruits
TRAF3, which interacts with kinases that mediate IRF3 (IFN regulatory factor 3) phosphorylation. IRF3 then translocates into the nucleus and regulates transcription of type one IFNs. Therefore, western blot analysis of IRF3 was performed and revealed a significant increase in SW-treated wild types compared with untreated controls and TLR32/2 mice with and without treatment (CTR 46.13+14.49 vs. SWT 102.94 +12.30; TLR32/2 24.48 +4.09 vs. TLR32/2 + SWT 10.63+3.25, P . 0.05; SWT vs. TLR32/2 + SWT, P , 0.01) (Figure 6G).
These data confirm that SW treatment solely induces TLR3
signalling. The TLR4 pathway is not influenced by this treatment.
4. Discussion
SWT has been shown to be effective in chronic and acute myocardial ischaemia. It was found to reduce angina symptoms and to improve left ventricular ejection fraction in patients suffering from coronary artery disease.9,10 As a mechanistic mode of action, the induction of angiogenesis was observed.8,29 However, the exact mechanism of how the
mechanical stimulus is translated into a biological response, the socalled mechanotransduction, remains unknown.
In the present work, we hypothesized that SWs cause a release of cytoplasmic RNA and thereby stimulate TLR3. As all cells in general contain high amounts of RNAs in the cytoplasm, this mechanism seems reasonable. Many of these RNAs seem to not code for specific proteins. Moreover, highly active cells such as myocytes or cardiomyocytes even contain higher amounts of RNA, making these cells particularly susceptible to the proposed mechanism. TLR3 is part of the innate immune system and has been described to be involved in angiogenesis. On one hand, TLR3 has been shown to
suppress angiogenesis;30,31 on the other hand, it has been described to induce VEGF-mediated angiogenesis in vitro and in vivo. 20,32 Finally, TLR3 was shown to have a protective effect on the arterial wall in early
stages of atherosclerosis.33 The resulting effect of TLR3 activation
might be dependent on the underlying pathologic condition in which
it modulates inflammation.
Inflammation-induced angiogenesis plays a pivotal role in noninfectious tissue regeneration and repair.34 In the present work, we
propose that the innate immune system, namely Toll-like receptor 3,
is an essential part of tissue repair and regeneration. This innate regenerative system can be stimulated or enhanced by low-energy SW
treatment.
In our experiments, we observed that SW treatment in acute hind limb ischaemia resulted in a significant increase of angiogenic response. Blood perfusion was restored and limb necrosis diminished. Effects were abolished in TLR3 knockout mice. Our data demonstrate that SWs stimulate TLR3 by the release of RNA. Inhibition of protein biosynthesis did not abolish observed effects. TLR3 and its readout gene IFN-b1 have been significantly upregulated in SW-treated endothelial cells. However, when TLR3 was silenced, the effect on IFN-b1 was completely lost. This finding indicates that the effects of SWs are mediated in a TLR3-dependent fashion. Additionally, the supernatant of TLR3-treated cells showed a marked increase in total RNA content. The same was true for serum of treated animals. This mechanistic finding is noteworthy, as just
recently heat shock protein 27 has been described to mediate
angiogenesis via TLR3 as well.20 However, when isolating RNA from the supernatant of treated and untreated cells and incubating it with another cells, no significant difference in TLR3 activation could be observed. We therefore hypothesized that RNA may form complexes with any protein and performed experiments with Proteinase K additional to RNase and indeed TLR3 signaling upon SWT was almost completely abolished. This indicates that not naked RNA, but RNA – protein complexes are responsible for the observed effect. One other possible mechanism for RNA release would be via exosomes as described earlier in another context.35 Our in vitro findings of delayed TLR3 stimulation 6 h after SW treatment are in line with the hypothesis that some kind of active mechanism is involved in RNA release or uptake, which stimulates TLR3 in contrast to direct stimulation
of TLR3 by its agonist Poly(I:C). We have been able to gain some array data that indicate miRNAs to be involved. However, whether this is the sole mechanism or other RNAs like long non-coding RNAs are involved as well is still a matter of investigation and future work will shed more light to the mechanism of TLR3 activation upon shockwave
treatment. TLR3 but not TLR4 signalling pathway was found to be involved after SW treatment of ischaemic muscle in vivo, as shown by IFN regulatory factor 3 (IRF3) phosphorylation. IRF3 translocates into the nucleus and
regulates transcription of type 1 IFNs and inflammatory cytokines. As these effects are abolished in TLR32/2 mice, we conclude that TLR3 plays a major role in SWT mechanotransduction. Summarizing, we found that the earlier described angiogenic effects of low-energy SW treatment in ischaemic muscle are mediated by TLR3 signalling. Nearly a complete loss of response has been observed in TLR3 knockout mice and TLR3-silenced cells. TLR3 stimulation
seems to be due to a release of cytosolic RNA into the extracellular space and to circulation subsequently. However, the exact mechanism of transmembrane trafficking of RNA after SW treatment as well as the specific type of RNA involved in TLR3 activation still remain to be elucidated in future experiments.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Acknowledgements
We are grateful to Prof. Shizuo Akira from Osaka University, Japan, for providing us FTO for the use of the TLR32/2 mice.
Conflict of interest: none declared.
Funding
This work was funded by a research grant from Bayer Pharma AG (Grants4Targets) to P.P. and by a research grant from Tissue Regeneration Technologies (TRT) Llc. to J.H. The sponsors of this study had no role in study design, data collection, analysis, and decision to publish.
References
- Gheorghiade M, Bonow RO. Chronic heart failure in the united states: a manifestation
of coronary artery disease. Circulation 1998;97:282 –289. - Marwick TH, Zuchowski C, Lauer MS, Secknus MA, Williams J, Lytle BW. Functional
status and quality of life in patients with heart failure undergoing coronary bypass surgery after assessment of myocardial viability. J Am Coll Cardiol 1999;33:750 758. - Hiatt WR. Medical treatment of peripheral arterial disease and claudication. N Engl J Med 2001;344:1608 –1621.
- Gupta R, Tongers J, Losordo DW. Human studies of angiogenic gene therapy. Circ Res 2009;105:724 –736.
- Sanganalmath SK, Bolli R. Cell therapy for heart failure: a comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ Res 2013;113:810 –834.
- Tepeko¨ylu¨ C, Wang FS, Kozaryn R, Albrecht-Schgoer K, Theurl M, Schaden W, Ke HJ, Yang Y, Kirchmair R, Grimm M, Wang CJ, Holfeld J. Shock wave treatment induces angiogenesis and mobilizes endogenous CD31/CD34-positive endothelial cells in a hind limb ischemia model: implications for angiogenesis and vasculogenesis. J Thorac Cardiovasc Surg 2013;146:971 – 978.
- Stojadinovic A, Elster EA, Anam K, Tadaki D, Amare M, Zins S, Davis TA. Angiogenic
response to extracorporeal shock wave treatment in murine skin isografts. Angiogenesis
2008;11:369 –380. - Holfeld J, Zimpfer D, Albrecht-Schgoer K, Stojadinovic A, Paulus P, Dumfarth J,
Thomas A, Lobenwein D, Tepeko¨ylu¨ C, Rosenhek R, Schaden W, Kirchmair R,
Aharinejad S, Grimm M. Epicardial shock-wave therapy improves ventricular function
in a porcine model of ischaemic heart disease. J Tissue Eng Regen Med 2014; doi:
10.1002/term.1890. - Khattab AA, Brodersen B, Schuermann-Kuchenbrandt D, Beurich H, To¨lg R, Geist V,
Scha¨fer T, Richardt G. Extracorporeal cardiac shock wave therapy: first experience
in the everyday practice for treatment of chronic refractory angina pectoris. Int J Cardiol
2007;121:84– 85. - Yang P, Guo T, Wang W, Peng YZ, Wang Y, Zhou P, Luo ZL, Cai HY, Zhao L, Yang HW.
Randomized and double-blind controlled clinical trial of extracorporeal cardiac shock
wave therapy for coronary heart disease. Heart Vessels 2013;28:284 –291. Aicher A, Heeschen C, Sasaki K, Urbich C, Zeiher AM, Dimmeler S. Low-energy shock - wave for enhancing recruitment of endothelial progenitor cells: a new modality to increase efficacy of cell therapy in chronic hind limb ischemia. Circulation 2006;114: 2823 –2830.
- Assmus B, Walter DH, Seeger FH, Leistner DM, Steiner J, Ziegler I, Lutz A, Khaled W, Klotsche J, Tonn T, Dimmeler S, Zeiher AM. Effect of shock wave-facilitated intracoronary cell therapy on LVEF in patients with chronic heart failure: the CELLWAVE randomized clinical trial. JAMA 2013;309:1622 – 1631.
- Wang FS, Wang CJ, Huang HJ, Chung H, Chen RF, Yang KD. Physical shock wave mediates membrane hyperpolarization and Ras activation for osteogenesis in human bone marrow stromal cells. Biochem Biophys Res Commun 2001;287:648–655.
- Holfeld J, Tepeko¨ylu¨ C, Kozaryn R, Urbschat A, Zacharowski K, Grimm M, Paulus P.Shockwave therapy differentially stimulates endothelial cells: implications on the control of inflammation via toll-like receptor 3. Inflammation 2014;37:65 –70.
- Kariko´ K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 2004;279:12542 –12550.
- Fabbri M, Paone A, Calore F, Galli R, Croce CM. A new role for microRNAs, as ligandsof Toll-like receptors. RNA Biol 2013;10:169 –174.
- Kleinman ME, Kaneko H, Cho WG, Dridi S, Fowler BJ, Blandford AD, Albuquerque RJ,Hirano Y, Terasaki H, Kondo M, Fujita T, Ambati BK, Tarallo V, Gelfand BD,Bogdanovich S, Baffi JZ, Ambati J. Short-interfering RNAs induce retinal degenerationvia TLR3 and IRF3. Mol Ther 2012;20:101 –108.
- Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med 2007; 13:460 – 469.
- Grelier A, Cras A, Balitrand N, Delmau C, Lecourt S, Lepelletier Y, Riesterer H, Freida D, Lataillade JJ, Lebousse-Kerdiles MC, Cuccini W, Peffault de Latour R, Marolleau JP, Uzan G, Larghero J, Vanneaux V. Toll-like receptor 3 regulates cord blood-derived endothelial cell function in vitro and in vivo. Angiogenesis 2013;16: 821– 836.
- Thuringer D, Jego G, Wettstein G, Terrier O, Cronier L, Yousfi N, He´brard S, Bouchot A, Hazoume´ A, Joly AL, Gleave M, Rosa-Calatrava M, Solary E, Garrido C. Extracellular HSP27 mediates angiogenesis through Toll-like receptor 3. FASEB J 2013;27:4169 –4183.
- Jackson JR, Seed MP, Kircher CH, Willoughby DA, Winkler JD. The codependence of
angiogenesis and chronic inflammation. FASEB J 1997;11:457 –465. - Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation
and repair after myocardial infarction. Circulation 2010;121:2437 –2445. - Couffinhal T, Silver M, Zheng LP, Kearney M, Witzenbichler B, Isner JM. Mouse model
of angiogenesis. Am J Pathol 1998;152:1667 –1679. - Schgoer W, Theurl M, Albrecht-Schgoer K, Jonach V, Koller B, Lener D, Franz WM,
Kirchmair R. Secretoneurin gene therapy improves blood flow in an ischemia model
in type 1 diabetic mice by enhancing therapeutic neovascularization. PLoS One 2013;
8:e74029. - Paulus P, Ockelmann P, Tacke S, Karnowski N, Ellinghaus P, Scheller B, Holfeld J,
Urbschat A, Zacharowski K. Deguelin attenuates reperfusion injury and improves outcome after orthotopic lung transplantation in the rat. PLoS One 2012;7:e39265. - Siow RC. Culture of human endothelial cells from umbilical veins. Methods Mol Biol
2012;806:265 – 274. - Messner B, Ploner C, Laufer G, Bernhard D. Cadmium activates a programmed, lysosomal membrane permeabilization-dependent necrosis pathway. Toxicol Lett 2012;212:
268 –275. - Paulus P, Stanley ER, Scha¨fer R, Abraham D, Aharinejad S. Colony-stimulating factor-1
antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer
Res 2006;66:4349 –4356. - Mittermayr R, Hartinger J, Antonic V, Meinl A, Pfeifer S, Stojadinovic A, Schaden W,
Redl H. Extracorporeal shock wave therapy (ESWT) minimizes ischemic tissue necrosis irrespective of application time and promotes tissue revascularization by stimulating
angiogenesis. Ann Surg 2011;253:1024 – 1032. - Cho WG, Albuquerque RJ, Kleinman ME, Tarallo V, Greco A, Nozaki M, Green MG,
Baffi JZ, Ambati BK, De Falco M, Alexander JS, Brunetti A, De Falco S, Ambati J. Small
interfering RNA-induced TLR3 activation inhibits blood and lymphatic vessel growth.
Proc Natl Acad Sci USA 2009;106:7137 –7142. - Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, Appukuttan B, Gibbs D, Yang Z, Kariko´ K, Ambati BK, Wilgus TA, DiPietro LA, Sakurai E, Zhang K, Smith JR, Taylor EW, Ambati J. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 2008;452:591 –597.
- Paone A, Galli R, Gabellini C, Lukashev D, Starace D, Gorlach A, De Cesaris P, Ziparo E, Del Bufalo D, Sitkovsky MV, Filippini A, Riccioli A. Toll-like receptor 3 regulates angiogenesis and apoptosis in prostate cancer cell lines through hypoxia-inducible factor 1 alpha. Neoplasia 2010;12:539 – 549.
- Cole JE, Navin TJ, Cross AJ, Goddard ME, Alexopoulou L, Mitra AT, Davies AH, Flavell RA, Feldmann M, Monaco C. Unexpected protective role for Toll-like receptor 3 in the arterial wall. Proc Natl Acad Sci USA 2011;108:2372 –2377.
- Frantz S, Vincent KA, Feron O, Kelly RA. Innate immunity and angiogenesis. Circ Res 2005;96:15–26.
- Sahoo S, Klychko E, Thorne T, Misener S, Schultz KM, Millay M, Ito A, Liu T, Kamide C, Agrawal H, Perlman H, Qin G, Kishore R, Losordo DW. Exosomes from human CD34( + ) cells mediate their proangiogenic paracrine activity. Circ Res 2011; 109:724 –728.
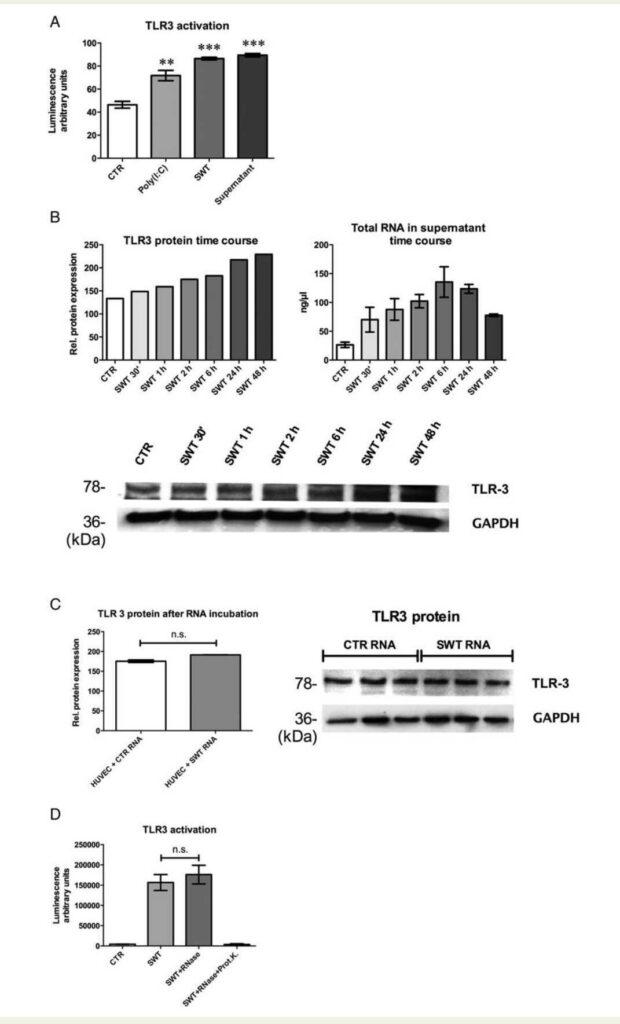
was detected as in directly treated cells and Poly(I:C)-stimulated cells (n ¼ 5, **P , 0.01, ***P , 0.001). (B) Confirmation of TLR3 stimulation at protein
level was confirmed by western blot analysis and showed a time-dependent result. In parallel, an increase of total RNA in supernatant over time could be
observed (n ¼ 3). (C) Incubation of HUVECs with isolated RNA from treated and untreated cells’ supernatant showed no significant difference in TLR3
protein. As naked RNA is unstable, we therefore hypothesized that RNA may be bound in a protein complex. (D) HEK reporter cells revealed that RNase
alone does not alter TLR3 stimulation upon SWT, but RNase in combination with proteinase K abolished the effect. This result states that RNA released
by SW-treated cells obviously is bound in a protein complex.
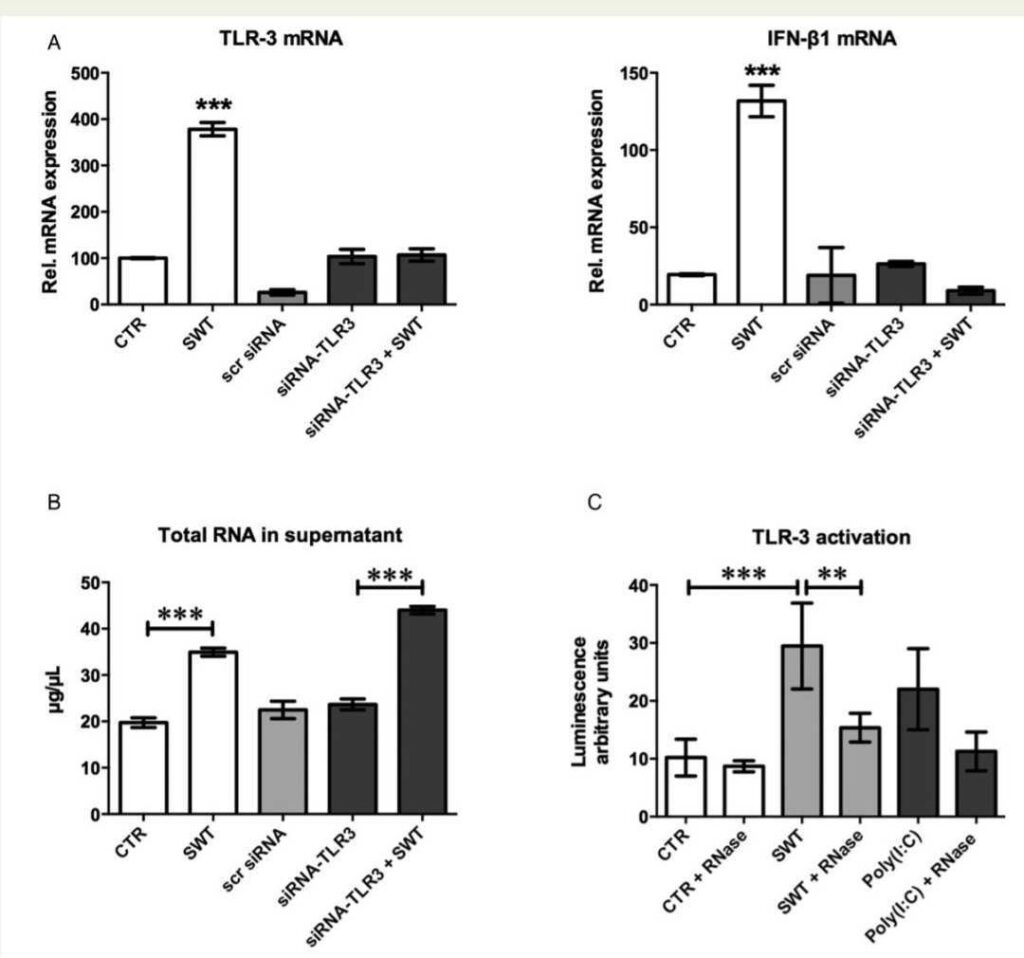
specific anti-TLR3 siRNA. It resulted in a loss of TLR3 response to SWT, which first confirms the efficacy of the knockdown and secondly indicates the
dependence of TLR3 in the transduction of SW responses. IFN-b1 served as a readout gene of downstream TLR3 signalling. There was also a loss of
IFN-b1 mRNA expression response after SWT in TLR3-silenced cells (n ¼ 3, ***P , 0.001 vs. all other groups). The use of scrambled siRNA confirmed
that transfection itself did not alter TLR3 or IFN-b1 expression. (B) SWT causes RNA release into supernatant. SW-treated HUVECs release increased
amounts of total RNA content into supernatant compared with their untreated controls. The same effect could be observed in TLR3-silenced cells
indicating that at least the release of mRNA to the extracellular space is independent from TLR3 (n ¼ 6, ***P , 0.001). (C) RNase abolishes the effect
of SWT. To verify whether the effect of supernatant from SW-treated endothelial cells is indeed mediated by cellular RNA, we incubated the supernatant
of SW-treated HUVECs with RNase and treated HEK TLR3 reporter cells therewith. The effect on TLR3 activation was almost completely abolished.
Poly(I:C) served as a positive control (n ¼ 4, **P , 0.01, ***P , 0.001).
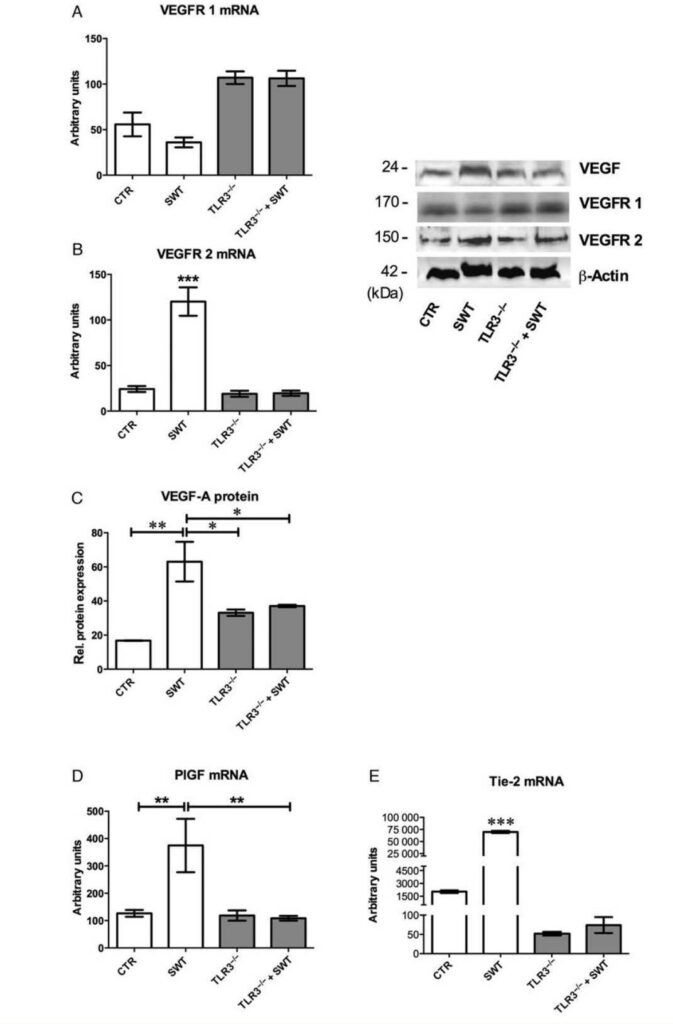
SWT-induced angiogenesis. VEGF receptor 1 is not increased by SW treatment (n ¼ 6). (B) Increase of VEGF receptor 2 by SWT. VEGFR 2 mRNA
shows a highly significant up-regulation in the treatment group (SWT) compared with wild-type controls and TLR32/2 mice (n ¼ 6, ***P , 0.001
vs. all other groups). This indicates that VEGFR 2 may be the pivotal receptor for VEGF-mediated angiogenesis following SWT. (C) VEGF-A protein
expression is up-regulated after SWT. Additionally to VEGF receptor 2 increase, VEGF-A protein expression was also increased in SW-treated wild
types compared with untreated controls or TLR32/2 animals (n ¼ 6, *P , 0.05, **P , 0.01). (D) PlGF increase indicates arteriogenesis after SWT.
SW-treated wild types showed a significant increase of PlGF mRNA levels indicating that maturation of capillaries by means of arteriogenesis is also
stimulated by SWT (n ¼ 6, **P , 0.01). (E) Increased Tie-2 expression after SWT is missing in TLR32/2 mice. The expression of the endothelial
cell receptor Tie-2 gene was found to be increased in SW-treated wild types compared with untreated controls. This indicates that the angiopoietin/Tie-2 system may strongly be involved in the induction of angiogenesis after SWT. (n ¼ 6, ***P , 0.001 vs. all other groups).
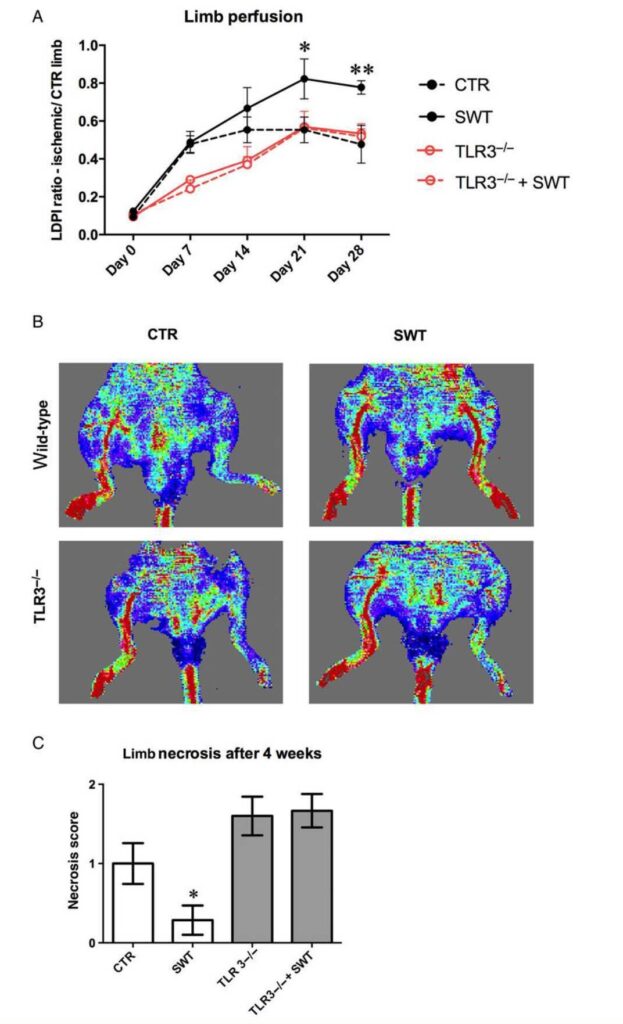
SW treatment nearly restored limb perfusion in wild-type mice after 28 days. This effect was abolished in TLR2/2 mice. Untreated wild-type controls and
treated or untreated TLR32/2 mice showed the same extent of self-regeneration. However, wild types regenerated at earlier time points (n ¼ 6, *P , 0.05,
**P , 0.01 vs. all other groups). (B) SWT improves blood perfusion in the hind limb ischaemia model. Representative pictures from Laser Doppler perfusion imaging reveal an almost complete restored blood perfusion in SW-treated wild types, whereas this effect was missing in SW-treated TLR32/2 mice.
(C) SWT decreases necrosis after hind limb ischaemia. SW-treated wild-type mice showed a significantly decreased necrosis score compared with untreated controls after 4 weeks. The amount of necrosis did not decrease after SW treatment in TLR32/2 mice. (n ¼ 6, *P , 0.05 vs. all other groups).
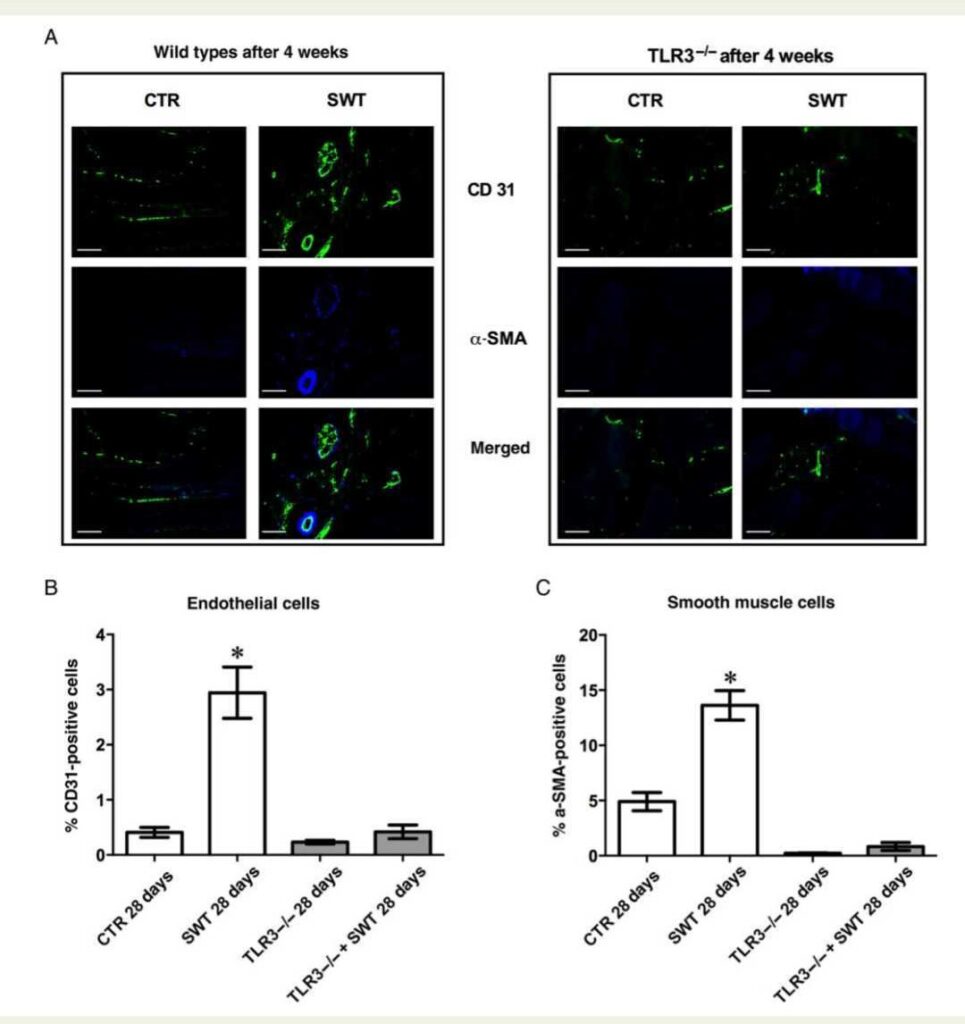
of CD31 for endothelial cells and a-SMA for smooth muscle cells in untreated wild-type controls (CTR) and after SW treatment (SWT). IF staining
clearly indicates that SW treatment induces angiogenesis and arteriogenesis in ischaemic muscle. Right panel shows the same staining in untreated
(CTR) and treated (SWT) TLR32/2 mice clearly depicting that SW effects are missing in ischaemic muscle lacking the TLR3 receptor (n ¼ 6, magnification ×400, calibration bar: 50 mm). (B) SWT increases endothelial cells in ischaemic muscles. Twenty-eight days after SWT higher numbers of endothelial cells have been detected in SW-treated muscle by IF staining indicating angiogenesis (n ¼ 6, *P , 0.05 vs. all other groups). Again effects were
abolished in TLR32/2 mice. (C) Smooth muscle cell recruitment is enhanced after SWT. Following SWT, a higher number of a-SMA-positive cells can be
found in ischaemic muscles of wild-type animals treated with SW. Together with the increased PlGF expression, these cells contribute to vessel maturation by means of arteriogenesis (n ¼ 6, *P , 0.05 vs. CTR and TLR32/2).
types, whereas this effect was abolished in TLR32/2 mice (n ¼ 6, ***P , 0.001 vs. all other groups). Increase of TLR3 downstream gene IFN-b1 mRNA
72 h after SWT confirms TLR3 stimulation. Effects are abolished in controls and TLR32/2 mice (n ¼ 6, ***P , 0.001 vs. all other groups). (B) Increase of
circulating RNA upon SWT. Circulating RNA in the serum of SW-treated wild-type and TLR2/2 animals was significantly increased (n ¼ 6, *P , 0.05,
***P , 0.001). Moreover, HUVECs incubated with serum for 6 h showed significant increase of TLR3 and downstream gene IFN-b1 mRNA. This effect
could be abolished adding RNase. (n ¼ 3, **P , 0.01 vs. all other groups). (C) IF of TLR3. IF shows TLR3 in ischaemic muscle of controls and SW-treated
wild-type mice after 72 h (magnification ×400, calibration bar: 50 mm; n ¼ 6, *P , 0.05). (D) Schematic drawing of TLR3 and TLR4 pathways. TLR3 and
TLR4 signalling both involve TRIF. TLR3 acts via IRF3 and TRAF6 activation, whereas TLR4 acts via MyD88 signalling as well. (E) Response TLR4 signalling
both involve TRIF. TLR3 acts via IRF3 to TLR3 activation is mediated by TRIF. TRIF protein is increased in SW-treated wild types but not in TLR2/2 mice.
(n ¼ 6, *P , 0.05, ***P , 0.001). (F) TRAF6 mediates TLR signalling. TRAF6 is utilized in TLR3 and TLR4 pathways. Increase of TRAF6 mRNA could be
observed in SW-treated wild types but was missing in controls and TLR32/2 mice. In the TLR4 pathway, TRAF6 is IRAK4 dependent, which was not
elevated (n ¼ 6, ***P , 0.001 vs. all other groups). (G) Increase of transcription regulator IRF3. IRF3 translocates into the nucleus and regulates transcription of IFN-b1. IRF3 protein was increased in SW-treated wild types but missing in controls and in TLR32/2 mice (n ¼ 6, *P , 0.05, **P , 0.01).
Johannes Holfeld1, Can Tepeko¨ ylu¨ 1, Christin Reissig2, Daniela Lobenwein1, Bertram Scheller2, Elke Kirchmair1, Radoslaw Kozaryn1, Karin Albrecht-Schgoer3, Christoph Krapf1, Karin Zins4, Anja Urbschat5, Kai Zacharowski2, Michael Grimm1, Rudolf Kirchmair3, and Patrick Paulus6*
1Department of Cardiac Surgery, Innsbruck Medical University, Innsbruck, Austria; 2Department of Anesthesiology, Intensive Care Medicine and Pain Therapy, University Hospital Frankfurt, Frankfurt am Main, Germany; 3Department of Internal Medicine III, Innsbruck Medical University, Innsbruck, Austria; 4Department of Anatomy and Cell Biology, Vienna Medical University, Vienna, Austria; 5Faculty of Medicine, Philipps University Marburg, Marburg, Germany; and 6Department of Anesthesiology and Operative Intensive Care Medicine, Kepler University Hospital,Linz, Austria
Received 16 February 2015; revised 6 November 2015; accepted 18 November 2015; online publish-ahead-of-print 16 December 2015