Shock Wave Therapy Improves Cardiac Function in a Model of Chronic Ischemic Heart Failure: Evidence for a Mechanism Involving VEGF Signaling and the Extracellular Matrix
Background
Mechanical stimulation of acute ischemic myocardium by shock wave therapy (SWT) is known to improve cardiac function by induction of angiogenesis. However, SWT in chronic heart failure is poorly understood. We aimed to study whether mechanical stimulation upon SWT improves heart function in chronic ischemic heart failure by induction of angiogenesis and postnatal vasculogenesis and to dissect underlying mechanisms.
Methods and Results
SWT was applied in a mouse model of chronic myocardial ischemia. To study effects of SWT on postnatal vasculogenesis, wild‐type mice received bone marrow transplantation from green fluorescence protein donor mice. Underlying mechanisms were elucidated in vitro in endothelial cells and murine aortic rings. Echocardiography and pressure/volume measurements revealed improved left ventricular ejection fraction, myocardial contractility, and diastolic function and decreased myocardial fibrosis after treatment. Concomitantly, numbers of capillaries and arterioles were increased. SWT resulted in enhanced expression of the chemoattractant stromal cell–derived factor 1 in ischemic myocardium and serum. Treatment induced recruitment of bone marrow–derived endothelial cells to the site of injury. In vitro, SWT resulted in endothelial cell proliferation, enhanced survival, and capillary sprouting. The effects were vascular endothelial growth factor receptor 2 and heparan sulfate proteoglycan dependent.
Conclusions
SWT positively affects heart function in chronic ischemic heart failure by induction of angiogenesis and postnatal vasculogenesis. SWT upregulated pivotal angiogenic and vasculogenic factors in the myocardium in vivo and induced proliferative and anti‐apoptotic effects on endothelial cells in vitro. Mechanistically, these effects depend on vascular endothelial growth factor signaling and heparan sulfate proteoglycans. SWT is a promising treatment option for regeneration of ischemic myocardium.
Clinical Perspective
What Is New?
- Shock wave therapy (SWT) improves cardiac function in a murine model of chronic ischemic cardiomyopathy by induction of angiogenesis and vasculogenesis and decreases myocardial fibrosis.
-
Vascular endothelial growth factor release from heparan sulfate proteoglycans results in enhanced endothelial cell proliferation, enhanced survival, and capillary sprouting.
-
Shock wave therapy induces enhanced expression of the chemoattractant stromal cell–derived factor 1 in ischemic myocardium and serum and results in higher numbers of bone marrow–derived endothelial cells at the site of injury.
What Are the Clinical Implications?
- Shock wave therapy develops a promising regenerative treatment option for patients with ischemic cardiomyopathy. It could serve as a promotor of “endogenous cell therapy” without exhibiting the disadvantages of cell harvesting necessary for conventional cell therapy.
-
In more than 3 decades of medical application, no severe side effects have been described.
-
Thus, shock wave therapy for regeneration of ischemic myocardium could be translated into a clinical setting efficiently.
Ischemic heart disease remains the leading cause of mortality (12.7%) worldwide, causing more than 7 million deaths per year.1
Infarction of the myocardium causes loss of cardiomyocytes. Remodeling of the infarction zone results in replacement of the necrotic myocardium by fibrotic scar tissue without contractile function. Besides a necrotic infarction core zone, chronically ischemic myocardium persists at the border zone of the infarction.2 Native angiogenesis after infarction most often is not sufficient for appropriate supply of hibernating cardiomyocytes, leading to pathologic left ventricle (LV) remodeling and ischemic cardiomyopathy, a clinical entity characterized by a poor prognosis and severe symptoms in affected patients.3 Management of this disease often is limited, and curative strategies beside heart transplantation are lacking. Therefore, therapies enhancing vascularization in the infarction zone by induction of capillary sprouting from existing vessels (angiogenesis) and/or by recruitment of bone marrow–derived endothelial cells (BMEC) for de novo vessel formation (vasculogenesis) are therefore of high relevance.4 For this purpose, strategies including cell therapy, gene therapy, and growth factor administration have been investigated.6 Although some clinical benefits have been reported, because of divergent results and safety concerns, none of the described methods has attained broad routine clinical use.7 Enhancing vascularization of chronically ischemic myocardium remains especially challenging, as production of growth factors for angiogenesis and chemoattractants for vasculogenesis are reduced in chronic fibrotic scars.
Shock waves are mechanical pressure waves with a specific physical wave profile used in clinical medicine for >30 years.9 At low energy levels, shock wave therapy (SWT) is in routine clinical use for the treatment of wound‐healing disturbances,10 tendinopathies, and nonhealing bone fractures.13 The regenerative effects of SWT have been mainly attributed to the induction of angiogenesis via growth factor release.16 Therefore, extensive research has been performed on cardiac SWT. In preclinical experiments, shock wave–treated hearts showed an increased number of capillaries and arterioles in the infarction border zone, thereby leading to a significantly improved left ventricular ejection fraction.20 In clinical studies, SWT caused the improvement of angina symptoms in patients with ischemic heart disease.23
Moreover, the combined treatment of SWT with subsequent systemic injection of endothelial progenitor cells in human patients with infarction has been investigated and showed convincing results by means of left ventricular ejection fraction improvement.25 In an experimental model of hindlimb ischemia, we showed that even endogenous endothelial progenitor cells become mobilized to systemic circulation upon SWT.26 However, their origin has not been identified so far.
In the present study we hypothesized that besides angiogenesis, cardiac SWT also induces the recruitment of BMECs to chronically ischemic myocardium. We propose involvement of heparan sulfate proteoglycan (HSPG)–bound growth factors in the extracellular matrix after the physical stimulus of SWT as an underlying mechanism. Angiogenic growth factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) are bound to the heparin‐binding domain of HSPGs in the extracellular matrix (ECM).28 The polysaccharides composed of a core protein and glycosamine chains have been described as mechanosensors orchestrating pivotal cellular functions upon mechanical force or tissue injury by growth factor release and subsequent induction of angiogenesis and tissue repair.30 In particular, the isoform VEGF165 is bound to HSPGs on the cellular surface.31
We aimed to analyze whether SWT would improve heart function by induction of angiogenesis and vasculogenesis in a model of chronic ischemic heart disease.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Animals
All procedures involving animals were approved by the national Animal Care and Use Committee, file number: BMWFW‐66.011/0138‐WF/V/3b/2014. Surgical procedures and animal care were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, volume 25, number 28, revised 1996), EU Directive 86/609 EEC, and Austrian Protection of Animals Act.
Investigators blinded to the treatment of the animals performed measurements and analysis. Twelve‐ to 14‐week‐old male C57BL/6 wild‐type mice (Charles River, Sulzfeld, Germany) weighing 25 to 30 g housed in the biomedical research facility of the Medical University of Innsbruck were randomly divided into treatment groups. During the experiments, mice were housed under standard conditions with a 12‐hour light/dark cycle. Water and commercial mouse diet were available ad libitum.
Myocardial Infarction Model
Anesthesia was administered by an intraperitoneal injection of ketamine hydrochloride 80 mg/kg (Graeub, Bern, Switzerland) and xylazine hydrochloride 5 mg/kg (aniMedica, Senden‐Bosensell, Germany). After endotracheal intubation and mechanical ventilation, the heart was exposed through a thoracotomy at the fourth intercostal space. Myocardial infarction was induced as described previously.22
SW Treatment
Animals received SWT 3 weeks after left anterior descending (LAD) ligation in a setting of chronic myocardial ischemia under anesthesia to the area of the anterior wall. Common ultrasound gel was used for coupling. The commercially available Orthogold device (TRT LLC, Tissue Regeneration Technologies, Woodstock, GA) served as shock wave source. For the in vivo application, 300 impulses were delivered to the ischemic area with an energy flux density of 0.38 mJ/mm2 at a frequency of 4 Hz. However, for all in vitro experiments 250 impulses with an energy flux density of 0.1 mJ/mm2 at a frequency of 3 Hz were used. The rationale of the treatment parameters is our experience from previous studies.32 At this low‐energy level, no adverse effects were observed. For the application of SWT to cultured cells, a specifically designed water bath was used as described previously.32
Bone Marrow Transplantation Model
The bone marrow transplantation model was performed as described previously.33 Briefly, sublethally irradiated C57BL/6 wild‐type mice received bone marrow cells (5 × 106) (bone marrow transplantation) from transgenic green fluorescence protein (GFP) mice (C57BL/6Tg(CAG‐EGFP)1Osb/J) by tail vein injection (n=6 per group). Four weeks after bone marrow transplantation, myocardial infarction was induced by LAD ligation. The treatment group received SWT 3 weeks after infarction, whereas control animals underwent sham treatment. Hearts were harvested 4 weeks after therapy. For identification of BMECs, mice were injected with 100 μL rhodamine‐conjugated BS‐1 lectin (Vectorlabs, Burlingame, CA) 20 minutes before euthanasia. Hearts were subsequently embedded in OCT compound (TISSUE TEK, Sakura Finetek, Staufen, Germany) and processed to 5‐μm‐thick sections. Samples were analyzed for red (rhodamine) labeled endothelial cells and for green (GFP) labeled bone marrow–derived cells with a Leica SP5 confocal microscope (Leica, Wetzlar, Germany; lens: HCX PL APO CS 63×1.2 [glycerine]). BMECs are expressed as number of double positive cells per high‐power field (HPF).
Echocardiography
Transthoracic echocardiography was performed using standard protocols for the assessment of heart function and morphometry. Briefly, lightly anaesthetized mice (0.5% isoflurane and 99.5% O2) were placed on a temperature‐controlled warming pad (kept at 37.5°C) and imaged in the supine position using a high‐resolution micro‐imaging system equipped with a 30‐MHz linear array transducer (Vevo770 Imaging System; VisualSonics Inc, Canada), respectively. Standard 2‐dimensional‐ and M‐mode tracings of the LV (long axis and short axis at papillary muscle level) were recorded and LV end‐diastolic diameter (LVEDD), LV end‐systolic diameter (LVESD), intraventricular septum thickness, and LV posterior wall thickness were averaged from 3 consecutive cardiac cycles under stable conditions. Fractional shortening was calculated using the equation: 100×((LVEDD−LVESD)/LVEDD). The investigator was blinded to the treatment.
Hemodynamic Pressure‐Volume Measurements
Invasive hemodynamic measurements and the analysis of pressure‐volume loops were performed as terminal procedure according to established protocols using a pressure‐volume conductance system (MPVS Ultra, Millar Instruments, USA) connected to the PowerLab 8/35 data acquisition system and analyzed using LabChart7pro software system (both ADInstruments, Sydney, Australia). Mice were anesthetized (3% isoflurane and 96%–97% O2), intubated, and mechanically ventilated. The animals were placed on a temperature‐controlled heating platform and the core temperature was maintained at 37.5°C. Anesthesia was reduced and kept at 1.5% isoflurane and 98.5% O2. A polyethylene catheter was inserted into the right external jugular vein for saline calibration (10% NaCl) at the end of the experiment to calculate the parallel volume. The 1.4‐F pressure‐volume conductance catheter (SPR‐839, Millar, USA) was inserted into the right carotid artery and advanced into the ascending aorta. Aortic (systemic) blood pressure and heart rate were recorded after stabilization for 5 minutes. Then the catheter was advanced into the LV under pressure control through the aortic valve as described previously.34
Cell Culture
After written informed consent of patients was obtained, umbilical cords were obtained from cesarean sections at the Department for Gynecology for isolation of human umbilical vein endothelial cells (HUVECs). Permission was given by the ethics committee of Medical University of Innsbruck (No. UN4435). Isolation was performed as described elsewhere.36 Freshly isolated HUVECs were cultivated in endothelial cell basal medium (CC‐3156, Lonza, Walkersville, USA) supplemented with EGM‐2 SingleQuots supplements (CC‐4176, Lonza). Cells in passages p3‐p5 were used for experiments. Commercially available H9c2 cells were used in the migration experiment (ATCC, Teddington, UK). Cells were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal calf serum. All cell culture experiments were performed at least in triplicate.
Endothelial Cell Proliferation Assay
Endothelial cell proliferation after SWT was analyzed as described previously.33
Aortic Ring Assay
The aortic ring assay was performed as described previously.37
Apoptosis Assay
Cells were pretreated with inhibitors as described under Endothelial Cell Proliferation Assay, treated with SWT, and serum starved in serum‐free EBM‐2 medium for 18 hours.
Terminal deoxynucleotidyl transferase dUTP nick‐end labeling (TUNEL) assay was performed as recommended by the manufacturer (Roche, Rotkreuz, Switzerland). Cells positive for TUNEL staining and for 4′,diamidino‐2‐phenylindole (DAPI) staining were counted and results are expressed as percentage of TUNEL‐positive cells of all DAPI‐stained cells.33
Migration Assay
A modified Boyden chamber assay was performed as described previously.38
Cytokine Array
Supernatant from SW‐treated HUVECs was analyzed using a human cytokine array kit as suggested by the manufacturer (human cytokine array kit; R&D Systems, Minneapolis, MN). Pixel density of membranes was quantified using Image J software (NIH, Bethesda, MD).
Western Blotting
Western blotting was performed as described previously.39
Stromal Cell–Derived Factor‐1 Enzyme‐Linked Immunosorbent Assay
Stromal cell–derived factor‐1 (SDF‐1) levels were quantified in murine serum 72 hours after treatment (n=6). Enzyme‐linked immunosorbent assay was performed according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). Detection of the concentration was performed using a microplate reader. Data are shown in nanograms per milliliter.
Reverse Transcription Polymerase Chain Reaction
Total RNA was extracted from homogenized tissue using the RNAII Kit (Machery‐Nagel; Düren, Germany) according to the manufacturer′s instructions. Real‐time reverse transcription polymerase chain reaction for gene expression analysis was performed with the ABI PRISM 7500 Sequence Detection System (Applied Biosystems, Waltham, MA). Primers were bought as assays on demand (Applied Biosystems, Waltham, MA; primers listed below). The polymerase chain reaction was performed in a final volume of 25 μL containing 1 μL cDNA, 12.5 μL Master Mix (Applied Biosystems, Waltham, MA), 1 μL fluorogenic hybridization probe, 6 μL primer mix, and 5.5 μL distilled water. The amplification consisted of a 2‐step polymerase chain reaction (40 cycles; 15‐s denaturation step at 95°C and 1 minute annealing/extension step at 60°C). Specific gene expression was normalized to the housekeeping gene β actin given by the formula 2‐ΔCt. The result for the relative gene expression was calculated by the 2‐DDCt method. The mean Ct values were calculated from double determinations, and samples were considered negative if the Ct values exceeded 40 cycles. Primers are described in Data S1.
Immunofluorescence Staining
Immunofluorescence staining was performed as described previously.39
Masson‐Trichrome Staining
For histological analysis, grafts were fixed in 4% (vol/vol) buffered formaldehyde, dehydrated with graded ethanol series, and embedded in paraffin. Masson‐trichrome staining was performed as recommended by the manufacturer (Carl Roth, Karlsruhe, Germany). The area of fibrosis and total area of the LV were measured using ImageJ (NIH, Bethesda, MD). Values are shown as infarction size (ratio: LV fibrosis/total area LV) as described previously.40
Statistical Analysis
All results are expressed as mean±SEM. Data sets were analyzed for normal distribution using the Kolmogorov–Smirnov test. If samples were normally distributed, a Student t test was performed for comparison between 2 groups. In the case of nonparametric distribution, a Mann–Whitney test was performed.
Multiple groups were analyzed by 1‐way ANOVA test followed by Bonferroni’s Multiple Comparison Test to determine statistical significance. Probability values <0.05 were considered statistically significant. All experiments were repeated at least in triplicate.
For further details regarding materials and methods, see Data S1.
Results
Chronic Ischemic Heart Failure: Functional Improvement by SWT
C57BL/6 mice were treated with SWT in a chronic infarction model (3 weeks after LAD ligation, n=6 per group). Cardiac function was measured 4 weeks after treatment using transthoracic echocardiography and a pressure‐volume catheter. Hearts were harvested subsequently for histological analyses (Figure 1A). Transthoracic echocardiography showed significant deterioration of myocardial function in untreated animals (ejection fraction in %: baseline 38.67±2.58 versus 7 weeks 29.80±1.43; fractional shortening in %: baseline 24.33±0.99 versus 7 weeks 18.40±0.75), whereas myocardial function of treated animals remained preserved (ejection fraction in %: baseline 39.50±1.29 versus 7 weeks 35.20±0.49; fractional shortening in %: baseline 25.33±1.20 versus 7 weeks 21.60±1.08) (Figure 1C) (representative m‐mode images are shown in Figure 1D).
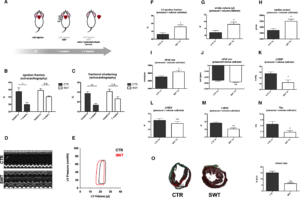
Cardiac function was evaluated additionally via invasive hemodynamic pressure‐volume measurement (representative pressure‐volume loops Figure 1E). Improvement of LV function and myocardial contractility by SWT was confirmed (ejection fraction in %: control [CTR] 26.25±2.14 versus SWT 35.80±2.577, P=0.029; stroke volume in μL: CTR 6.8±0.58 versus SWT 9.2±0.66, P=0.026; cardiac output in μL/min: CTR 3457±317.3 versus SWT 4987±361.6, P=0.018; dP/dt max in mm Hg/s: CTR 5508±157.9 versus SWT 6171±178.1, P=0.0043; dP/dt minute in mm Hg/s: CTR −5066±268.5 versus SWT −5588±236.2, P=0.19) (Figure 1F through 1J). SW‐treated animals showed improved diastolic function with enhanced isovolumetric relaxation compared with untreated CTRs (LV end diastolic pressure [LVEDP] in mm Hg: CTR 17.50±0.65 versus SWT 14.40±0.93, P=0.036; LV end diastolic volume in μL: CTR 25.75±1.18 versus SWT 23.80±1.28, P=0.31; LV end systolic volume in μL: CTR 19.00±0.71 versus SWT 15.20±0.73, P=0.0317; tau in ms: CTR 9.12±0.24 versus SWT 8.45±0.13, P=0.038) (Figure 1K through 1N). Masson‐trichrome staining of myocardial sections from shockwave‐treated and ‐untreated CTR mice revealed remarkably decreased infarction size (% of LV: CTR 15.07±1.39 versus SWT 6.17±0.72, P<0.0001) (Figure 1O).
Shockwave Therapy Induces Angiogenesis and Arteriogenesis and Upregulates Pivotal Angiogenic Factors In Vivo
Treated hearts were analyzed for angiogenic gene expression 72 hours after SWT via reverse transcription polymerase chain reaction. SWT resulted in increased mRNA levels of VEGF (relative gene expression [arbitrary units A.U.]: CTR 18.50±1.06 versus SWT 26.64±2.897, P=0.02) (Figure 2A), its pivotal receptor VEGFR2 (relative gene expression [A.U.]: CTR 19.50±2.01 versus SWT 28.90±2.850, P=0.027) (Figure 2B), placental growth factor (relative gene expression [A.U.]: CTR 0.52±0.043 versus SWT 0.78±0.07, P=0.006) (Figure 2C), and FGF (relative gene expression [A.U.]: CTR 0.94±0.11 versus SWT 1.47±0.09, P=0.009) (Figure 2D). While VEGF and FGF stimulate capillary sprouting by induction of endothelial proliferation and migration, placental growth factor induces maturation and stabilization of vessels contributing to arteriogenesis.
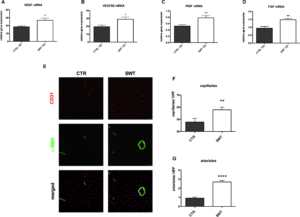
Vascularization in ischemic myocardium therefore was visualized by immunofluorescence staining for CD31 (endothelial cells, capillaries) and α‐smooth muscle actin, smooth muscle cells, and arterioles (Figure 2E). A higher blood vessel density was found in the peri‐infarct regions of shockwave‐treated animals compared with untreated controls. Significantly higher numbers of capillaries (Figure 2F) (capillaries per HPF: CTR 47.78±2.95 versus SWT 57.85±2.24, P=0.0342) as well as arterioles and arteries (arterioles per HPF: CTR 0.92±0.11 versus SWT 2.69±0.17, P<0.0001) (Figure 2G) were observed in the treatment group. These observations indicate induction of angiogenesis and arteriogenesis in the infarct border zone of chronically ischemic myocardium by SWT.
Shock‐Wave Effects on Endothelial Cells and Angiogenesis Ex Vivo
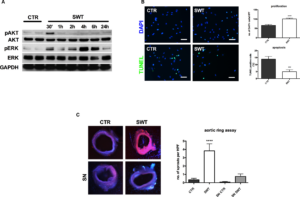
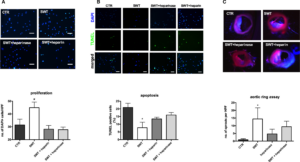
Two protein kinases play a crucial role during angiogenesis: activation of mitogen‐activated p42/44 ERK1/2 regulates endothelial cell proliferation, while activation of Akt/protein kinase B is required for remodeling of the cytoskeleton and concomitant cell migration. Both kinases are activated by phosphorylation. SWT of HUVECs resulted in phosphorylation of Akt after 30 minutes. ERK was activated by SWT after 30 minutes and, as a second peak, after 4 and 6 hours (Figure 3A). This resulted in induction of endothelial proliferation after SWT (number of DAPI‐positive cells/HPF: CTR 66.28±4.70 versus SWT 100.9±4.19, P<0.001) (Figure 3B). To investigate SW effects on cell survival, HUVECs were starved, treated, and subsequently analyzed via TUNEL assay. We found an anti‐apoptotic effect of SWT (% of TUNEL‐positive cells: CTR 13.91±1.97 versus SWT 4.99±1.36, P=0.0006) (Figure 3B). To verify whether the observed effects do in fact lead to capillary sprouting, we performed an aortic ring assay. For this purpose, murine aortas were cultured in matrigel and treated with SWT, whereas controls remained untreated. After 7 days, capillary sprouts were quantified. SWT induced significantly increased capillary sprouting compared with untreated controls (number of sprouts/HPF: CTR 0.36±0.17 versus SWT 3.83±0.85, P<0.0001). Interestingly, treatment of aortic rings with supernatant from SW‐treated cells improved capillary sprouting as well (number of sprouts/HPF: CTR 0.071±0.071 versus SWT 0.7500±0.31, P=0.0155) (Figure 3C). These results clearly indicate that SWT exhibits a potent angiogenic effect in endothelial cells via phosphorylation of Akt and ERK, induction of proliferation, inhibition of apoptosis, as well as enhanced capillary sprouting in an ex vivo angiogenesis model.
Shock Wave Effects Depend on VEGF Signaling
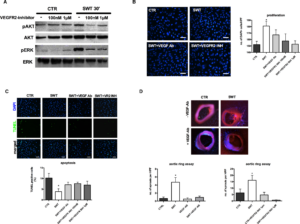
VEGF signaling has been shown to play a major role in SW‐induced angiogenesis. We aimed to clarify whether observed effects were in fact VEGF dependent and whether VEGFR2 stimulation and consecutive MAPK activation were responsible for angiogenesis after treatment. Inhibition of VEGFR2 via vandetanib (100 nm and 1 μm) abolished phosphorylation of Akt and ERK 30 minutes after SWT, clearly indicating that SW‐induced activation of MAPK is VEGFR2 dependent (Figure 4A). Moreover, pretreatment of HUVECs with anti‐VEGF antibody (2 μg/mL) or VEGFR2 inhibitor vandetanib (100 nmol/L and 1 μmol/L) completely blocked the proliferative SW effect (number of DAPI‐positive cells/HPF: CTR 152.1±5.06, SWT 180.9±7.4, SWT+VEGF‐Ab 167.3±7.86, SWT+VEGFR2 inhibitor (INH) 100 nmol/L 157.7±11.14, SWT+VEGFR2 INH 1 μmol/L 152.8±5.22) (Figure 4B). SWT did not show any anti‐apoptotic effect in cells pretreated with anti‐VEGF antibody or VEGFR2 inhibitor (% of TUNEL‐positive cells: CTR 10.2±2.19, SWT 3.89±1.28, SWT+VEGF‐Ab 7.08±1.16, SWT+VEGFR2 INH 100 nmol/L 7.61±0.69, SWT+VEGFR2 INH 1 μmol/L 6.97±1.75) (Figure 4C). We found no significant capillary sprouting in the aortic ring assay upon VEGF/VEGFR2 inhibition after SWT (VEGF‐ab: number of sprouts/HPF: CTR 0.64±0.36, SWT 4.69±1.18, SWT+VEGF‐Ab 0.93±0.32, VEGF‐Ab 0.53±0.29; VEGFR2 INH: CTR 6.42±2.9, SWT 16.08±3.94, VEGFR2 INH 1 μmol/L 4.77±2.04, SWT+VEGFR2 INH 0.67±0.29) (Figure 4D). These results indicate clearly that the angiogenic effects of SWT are dependent on the VEGF/VEGFR2 pathway.
HSPG Bound Growth Factors Mediate SW Effects
The ECM contains growth factors from the VEGF and FGF families bound to HSPGs. Thus, growth factors can be provided rapidly in case of tissue injury for tissue repair. Prolonged ERK activation might indicate effects of HSPG‐bound growth factors in SWT stimulation. Therefore, heparin and heparinase were tested to influence SWT effects on endothelial cells and ex vivo angiogenesis. We pretreated HUVECs with heparinase (50 mU) or heparin (10 μg/mL) for growth factor depletion of the ECM.41 Pretreatment abolished proliferation upon SWT (number of DAPI‐positive cells/HPF: CTR 31.47±4.49, SWT 44.93±4.38, SWT+heparinase 27.8±1.96, SWT+heparin 28.13±2.86) (Figure 5A). SWT had no more effect on apoptosis after growth factor depletion (% of TUNEL‐positive cells: CTR 20.9±2.71, SWT 7.76±3.53, SWT+heparin 13.45±1.11, SWT+heparinase 15.98±1.67) (Figure 5B). We found no significant capillary sprouting in the ex vivo angiogenic ring assay after heparinase treatment (number of sprouts/HPF: CTR 0.86±0.71, SWT 14.4±7.27, heparinase 4.69±3.05, SWT+heparinase 9.39±3.58, P=0.107) (Figure 5C). These results indicate that HSPG‐bound growth factors are involved in angiogenesis after SWT.
SWT Stimulates Vasculogenesis in Chronic Ischemic Heart Failure
We aimed to address whether SWT stimulates vasculogenesis by recruitment of bone marrow–derived endothelial cells to chronically ischemic myocardium. In order to evaluate shockwave‐induced vasculogenesis in vivo, wild‐type mice received bone marrow transplantation from GFP donor mice (n=6 per group). Subsequently, animals underwent LAD ligation. After myocardial remodeling (3 weeks), chronically ischemic myocardium received SWT (Figure 6A). SWT enhanced incorporation of BMECs, as shown by increased numbers of cells double positive for rhodamine‐labeled isolectin (injected intravenously for staining of host vessels) and for GFP (representing bone marrow–derived cells after bone marrow transplantation). Examination of double‐positive cells per HPF revealed significantly higher numbers of BMECs in the SWT group compared with untreated controls (BMECs/HPF: CTR 3.98±0.6 versus SWT 17.89±1.59, P<0.0001) (Figure 6B, arrowheads).
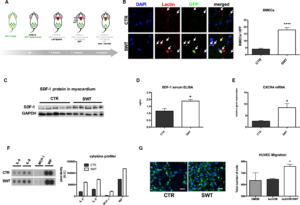
SDF‐1 serves as one of the most potent chemoattractants for the recruitment of endothelial cells from the bone marrow. To evaluate in vivo SDF‐1 levels after SWT Western blot, analysis of treated myocardium was performed from peri‐infarct samples (n=5) and it was confirmed that myocardial SDF‐1 protein levels were increased in the treatment group (Figure 6C). To analyze systemic chemotaxis upon SWT, we performed an SDF‐1 ELISA from blood serum 72 hours after treatment. We found higher levels of SDF‐1 in the circulation of treated animals (ng/mL: CTR 1.16±0.18 versus SWT 1.88±0.12, P=0.0102) (Figure 6D). In parallel, mRNA levels of the SDF‐1‐specific receptor C‐X‐C chemokine receptor type 4 were increased in treated myocardium (relative gene expression [A.U.]: CTR 2.58±0.31 versus SWT 8.47±1.71, P=0.043) (Figure 6E).
To elucidate effects of other chemoattractants after SWT in vitro, we treated HUVECs with SWT. Supernatant of treated cells contained higher protein amounts of interleukin (IL)‐6 (pixel density [A.U.]: CTR 19 002±768 versus SWT 59 973±2151), IL‐8 (pixel density [A.U.]: CTR 29 495.5±1506.5 versus SWT 71 890.5±2629.5), monocyte chemoattractant protein 1 (pixel density [A.U.]: CTR 2216±1537 versus SWT 20 076±2765), and macrophage migration inhibitory factor (pixel density [A.U.]: CTR 73 516±264 versus SWT 119 329±126), all crucial chemoattractants for cell recruitment (Figure 6F). Indeed, preconditioned medium of ischemic cardiomyocytes treated with SWT induced chemotaxis of endothelial cells in a modified Boyden chamber migration assay (number of cells/HPF: CTR 645.0±8.0 versus SWT 758.0±17.62, P=0.017) (Figure 6G).
Discussion
In previous publications, we demonstrated VEGF release and subsequent angiogenesis in acute models of hindlimb and myocardial ischemia after SWT.16 In daily clinical practice, however, chronic ischemic heart failure represents an even more challenging problem because of limited therapy and poor prognosis of this disease. The aims of this study therefore were to use a model of chronic ischemic heart disease and to investigate the pathophysiology of heart failure with and without SWT and additionally to dissect underlying mechanisms. Measurements by echocardiography as well as invasively by catheter showed improved systolic and diastolic heart function by SWT. In a porcine study, Uwatoku et al found improvement of cardiac function when applied directly after induction of myocardial infarction by preventing postinfarction remodeling. However, there was no beneficial SW effect on chronic ischemic myocardium in their setting.42 This discrepancy might be because of the divergent models of myocardial ischemia induction, because the group used excision of the circumflex artery, whereas in our model LAD ligation was performed.
Histology revealed decreased fibrosis, increased density of blood vessels, and recruitment of bone marrow–derived endothelial cells. In the chronic ischemic myocardium, pivotal angiogenic (VEGF and FGF) and vasculogenic factors (SDF‐1) as well as respective receptors were upregulated by SWT. Mechanistically, SWT stimulated endothelial cell proliferation, inhibited apoptosis, and activated intracellular protein kinases Akt and ERK, and inhibition of effects by blockade of VEGFR2 indicates an important role of VEGF signaling in SWT response. A late second peak of ERK activation after 4 hours prompted us to evaluate whether growth factors bound to HSPGs mediate effects of SWT.33
Currently it remains unknown how the mechanical stimulus of SWT stimulates VEGF signaling. A recent study showed crucial involvement of the mechanosensors caveolin‐1 and ß1‐integrin in SW‐induced mechanotransduction, because angiogenic response was clearly reduced after knock‐down of the proteins.43 However, because numerous immediate angiogenic effects of SWT have been described, we aimed to investigate the release of stored growth factors rather than de novo synthesis. The cellular VEGF reservoir is mainly found in the ECM bound to HSPGs.31 HSPGs were described as mechanosensors that are involved in orchestration of angiogenesis and endothelial cell migration. We therefore hypothesized that the mechanical stimulus of SWT leads to growth factor release or redistribution from HSPGs
facilitating binding of factors such as VEGF to its receptors. We confirmed that pretreatment of HUVECs with heparinase I and III for cleavage of heparin and heparin sulfate of HSPG and subsequent disruption of HSPG30 abolished SWT‐induced phosphorylation of Akt and ERK, endothelial cell proliferation, and enhanced survival and capillary sprouting. Although we did not find increased VEGF concentrations in supernatants of SWT‐treated cells (data not shown), it is conceivable that SWT induces redistribution of growth factors in the ECM, thus facilitating growth factor binding to the receptors. Neutralization of VEGF using a VEGF antibody antagonized angiogenic SW effects in vitro, as did inhibition of VEGFR2. In combination with our data of impaired SWT effects after heparinase, we therefore suggest that SWT induces angiogenesis via VEGF release/redistribution from extracellular HSPGs. Released VEGF binds to VEGFR2 and thus initiates angiogenesis. We already showed in a previous publication that SWT stimulates phosphorylation/activation of VEGFR2 without further cytokine stimulation.16
Moreover, we aimed to analyze whether the newly formed vessels after SWT originate from angiogenesis or whether postnatal vasculogenesis is also involved. We determined previously that SWT increases the number of CD34‐positive cells in the peripheral blood.26 Additionally, it was described that SWT enhances the recruitment of intravenously injected endothelial progenitor cells to ischemic hindlimbs.27 However, it was not yet known whether SW treatment also enhances the recruitment and homing of endogenous bone marrow–derived endothelial cells to the site of ischemic injury to induce de novo synthesis of blood vessels within chronic ischemic myocardium. To study this question, wild‐type mice received bone marrow transplantation from GFP donor mice, and underwent LAD ligation and SWT. BMECs could be identified as GFP+ endothelial cells within the myocardium. We found significantly more BMECs after SWT, showing for the first time that SWT does enhance recruitment and homing of autologous bone marrow–derived endothelial cells to chronic ischemic myocardium. SDF‐1, a pivotal chemoattractant for BMECs,27 was upregulated by SWT in the myocardium and in serum of treated animals as was the SDF receptor C‐X‐C chemokine receptor type 4 in myocardium. Additionally, we found enhanced expression of chemokines important for BMEC migration such as IL‐6, IL‐8, monocyte chemoattractant protein 1, and migration inhibitory factor after SWT. These data indicate that SWT applied to the chronic ischemic myocardium induces not only angiogenesis but also postnatal vasculogenesis.
Summarizing, we found improved cardiac function by SWT in a model of chronic ischemic heart failure as measured by echocardiography and invasively by catheter, detected reduction of fibrotic scar tissue, and improved neovascularization via angiogenesis and postnatal vasculogenesis. We were able to show that the mechanical impulse of SWT translates to a HSPG‐ and VEGF‐dependent stimulation of intracellular signal transduction pathways, increased proliferation and inhibition of apoptosis in vitro and stimulation of capillary sprouting ex vivo in aortic rings. Inhibition of these effects by heparinase and by blocking of VEGF and VEGFR2 indicates a pivotal role of VEGFR2 stimulated by ECM‐stored VEGF in SWT‐induced angiogenesis.
Recently, we demonstrated improved angiogenesis and subsequent improvement of heart function in a model of acute myocardial ischemia.22 However, in this study we were able to show for the first time improved vascularization in chronically ischemic myocardium and concomitant improvement of cardiac function. This is highly significant, because regenerative therapy options to improve heart function are limited for the increasing number of patients with chronic ischemic heart failure.
SWT could serve as a promotor of “endogenous cell therapy” without exhibiting the disadvantages of cell harvesting necessary for conventional cell therapy. In addition, long‐term effects of SWT are well known, because it has been used in medicine for more than 3 decades. No severe side effects were described after SWT. Thus, SWT for regeneration of ischemic myocardium could be translated into a clinical setting efficiently.44
Sources of Funding
This work was funded by a research grant from Tissue Regeneration Technologies Llc. to Holfeld and by a research grant from Bayer Pharma AG (Grants4Targets) to Paulus. The sponsors of this study had no role in study design, data collection, analysis, and decision to publish or prepare the manuscript.
Disclosures
Grimm and Holfeld are shareholders of the Medical University of Innsbruck spin‐off Heart Regeneration Technologies GmbH, a company that aims to develop direct cardiac shockwave devices for human use. The remaining authors have no disclosures to report.
Footnotes
*Correspondence to: Johannes Holfeld, MD, University Clinic of Cardiac Surgery, Innsbruck Medical University, Anichstraße 35, 6020 Innsbruck, Austria. E‐mail: johannes.
References
- Finegold JA, Asaria P, Francis DP. Mortality from ischaemic heart disease by country, region, and age: statistics from World Health Organisation and United Nations. Int J Cardiol. 2013; 168:934–945.CrossrefMedlineGoogle Scholar
- Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000; 101:2981–2988.LinkGoogle Scholar
- Casey TM, Arthur PG. Hibernation in noncontracting mammalian cardiomyocytes. Circulation. 2000; 102:3124–3129.LinkGoogle Scholar
- May D, Gilon D, Djonov V, Itin A, Lazarus A, Gordon O, Rosenberger C, Keshet E. Transgenic system for conditional induction and rescue of chronic myocardial hibernation provides insights into genomic programs of hibernation. Proc Natl Acad Sci USA. 2008; 105:282–287.CrossrefMedlineGoogle Scholar
- Ware JA, Simons M. Angiogenesis in ischemic heart disease. Nat Med. 1997; 3:158–164.CrossrefMedlineGoogle Scholar
- Zhang H, van Olden C, Sweeney D, Martin‐Rendon E. Blood vessel repair and regeneration in the ischaemic heart. Open Heart. 2014; 1:e000016.CrossrefMedlineGoogle Scholar
- Abbott A. Doubts over heart stem‐cell therapy. Nature. 2014; 509:15–16.CrossrefMedlineGoogle Scholar
- Check E. Cancer fears cast doubts on future of gene therapy. Nature. 2003; 421:678.CrossrefMedlineGoogle Scholar
- Chaussy C, Brendel W, Schmiedt E. Extracorporeally induced destruction of kidney stones by shock waves. Lancet. 1980; 2:1265–1268.CrossrefMedlineGoogle Scholar
- Ottomann C, Stojadinovic A, Lavin PT, Gannon FH, Heggeness MH, Thiele R, Schaden W, Hartmann B. Prospective randomized phase II trial of accelerated reepithelialization of superficial second‐degree burn wounds using extracorporeal shock wave therapy. Ann Surg. 2012; 255:23–29.CrossrefMedlineGoogle Scholar
- Schaden W, Thiele R, Kolpl C, Pusch M, Nissan A, Attinger CE, Maniscalco‐Theberge ME, Peoples GE, Elster EA, Stojadinovic A. Shock wave therapy for acute and chronic soft tissue wounds: a feasibility study. J Surg Res. 2007; 143:1–12.CrossrefMedlineGoogle Scholar
- Ottomann C, Hartmann B, Tyler J, Maier H, Thiele R, Schaden W, Stojadinovic A. Prospective randomized trial of accelerated re‐epithelization of skin graft donor sites using extracorporeal shock wave therapy. J Am Coll Surg. 2010; 211:361–367.CrossrefMedlineGoogle Scholar
- Wang CJ, Yang KD, Ko JY, Huang CC, Huang HY, Wang FS. The effects of shockwave on bone healing and systemic concentrations of nitric oxide (NO), TGF‐beta1, VEGF and BMP‐2 in long bone non‐unions. Nitric Oxide. 2009; 20:298–303.CrossrefMedlineGoogle Scholar
- Chen YJ, Kuo YR, Yang KD, Wang CJ, Sheen Chen SM, Huang HC, Yang YJ, Yi‐Chih S, Wang FS. Activation of extracellular signal‐regulated kinase (ERK) and p38 kinase in shock wave‐promoted bone formation of segmental defect in rats. Bone. 2004; 34:466–477.CrossrefMedlineGoogle Scholar
- Wang C‐J, Yang KD, Wang F‐S, Hsu C‐C, Chen H‐H. Shock wave treatment shows dose‐dependent enhancement of bone mass and bone strength after fracture of the femur. Bone. 2004; 34:225–230.CrossrefMedlineGoogle Scholar
- Holfeld J, Tepeköylü C, Blunder S, Lobenwein D, Kirchmair E, Dietl M, Kozaryn R, Lener D, Theurl M, Paulus P, Kirchmair R, Grimm M. Low energy shock wave therapy induces angiogenesis in acute hind‐limb ischemia via VEGF receptor 2 phosphorylation. PLoS One. 2014; 9:e103982.CrossrefMedlineGoogle Scholar
- Chen YJ, Wurtz T, Wang CJ, Kuo YR, Yang KD, Huang HC, Wang FS. Recruitment of mesenchymal cells and expression of TGF‐beta 1 and VEGF in the early stage of shock wave‐promoted bone regeneration of segmental defect in rats. J Orthop Res. 2004; 22:526–534.CrossrefMedlineGoogle Scholar
- Tepekoylu C, Lobenwein D, Urbschat A, Graber M, Pechriggl EJ, Fritsch H, Paulus P, Grimm M, Holfeld J. Shock wave treatment after hindlimb ischaemia results in increased perfusion and M2 macrophage presence. J Tissue Eng Regen Med. 2018; 12:e486–e494.CrossrefMedlineGoogle Scholar
- Weihs AM, Fuchs C, Teuschl AH, Hartinger J, Slezak P, Mittermayr R, Redl H, Junger WG, Sitte HH, Runzler D. Shock wave treatment enhances cell proliferation and improves wound healing by ATP release‐coupled extracellular signal‐regulated kinase (ERK) activation. J Biol Chem. 2014; 289:27090–27104.CrossrefMedlineGoogle Scholar
- Nishida T, Shimokawa H, Oi K, Tatewaki H, Uwatoku T, Abe K, Matsumoto Y, Kajihara N, Eto M, Matsuda T, Yasui H, Takeshita A, Sunagawa K. Extracorporeal cardiac shock wave therapy markedly ameliorates ischemia‐induced myocardial dysfunction in pigs in vivo. Circulation. 2004; 110:3055–3061.LinkGoogle Scholar
- Holfeld J, Zimpfer D, Albrecht‐Schgoer K, Stojadinovic A, Paulus P, Dumfarth J, Thomas A, Lobenwein D, Tepekoylu C, Rosenhek R, Schaden W, Kirchmair R, Aharinejad S, Grimm M. Epicardial shock‐wave therapy improves ventricular function in a porcine model of ischaemic heart disease. J Tissue Eng Regen Med. 2016; 10:1057–1064.CrossrefMedlineGoogle Scholar
- Tepekoylu C, Primessnig U, Polzl L, Graber M, Lobenwein D, Nagele F, Kirchmair E, Pechriggl E, Grimm M, Holfeld J. Shockwaves prevent from heart failure after acute myocardial ischaemia via RNA/protein complexes. J Cell Mol Med. 2017; 21:791–801.CrossrefMedlineGoogle Scholar
- Fukumoto Y, Ito A, Uwatoku T, Matoba T, Kishi T, Tanaka H, Takeshita A, Sunagawa K, Shimokawa H. Extracorporeal cardiac shock wave therapy ameliorates myocardial ischemia in patients with severe coronary artery disease. Coron Artery Dis. 2006; 17:63–70.CrossrefMedlineGoogle Scholar
- Kikuchi Y, Ito K, Ito Y, Shiroto T, Tsuburaya R, Aizawa K, Hao K, Fukumoto Y, Takahashi J, Takeda M, Nakayama M, Yasuda S, Kuriyama S, Tsuji I, Shimokawa H. Double‐blind and placebo‐controlled study of the effectiveness and safety of extracorporeal cardiac shock wave therapy for severe angina pectoris. Circ J. 2010; 74:589–591.CrossrefMedlineGoogle Scholar
- Assmus B, Walter DH, Seeger FH, Leistner DM, Steiner J, Ziegler I, Lutz A, Khaled W, Klotsche J, Tonn T, Dimmeler S, Zeiher AM. Effect of shock wave‐facilitated intracoronary cell therapy on LVEF in patients with chronic heart failure: the CELLWAVE randomized clinical trial. JAMA. 2013; 309:1622–1631.CrossrefMedlineGoogle Scholar
- Tepekoylu C, Wang FS, Kozaryn R, Albrecht‐Schgoer K, Theurl M, Schaden W, Ke HJ, Yang Y, Kirchmair R, Grimm M, Wang CJ, Holfeld J. Shock wave treatment induces angiogenesis and mobilizes endogenous CD31/CD34‐positive endothelial cells in a hindlimb ischemia model: implications for angiogenesis and vasculogenesis. J Thorac Cardiovasc Surg. 2013; 146:971–978.CrossrefMedlineGoogle Scholar
- Aicher A, Heeschen C, Sasaki K, Urbich C, Zeiher AM, Dimmeler S. Low‐energy shock wave for enhancing recruitment of endothelial progenitor cells: a new modality to increase efficacy of cell therapy in chronic hind limb ischemia. Circulation. 2006; 114:2823–2830.LinkGoogle Scholar
- Iozzo RV, San Antonio JD. Heparan sulfate proteoglycans: heavy hitters in the angiogenesis arena. J Clin Invest. 2001; 108:349–355.CrossrefMedlineGoogle Scholar
- Martino MM, Briquez PS, Ranga A, Lutolf MP, Hubbell JA. Heparin‐binding domain of fibrin(ogen) binds growth factors and promotes tissue repair when incorporated within a synthetic matrix. Proc Natl Acad Sci USA. 2013; 110:4563–4568.CrossrefMedlineGoogle Scholar
- Moon JJ, Matsumoto M, Patel S, Lee L, Guan JL, Li S. Role of cell surface heparan sulfate proteoglycans in endothelial cell migration and mechanotransduction. J Cell Physiol. 2005; 203:166–176.CrossrefMedlineGoogle Scholar
- Beckouche N, Bignon M, Lelarge V, Mathivet T, Pichol‐Thievend C, Berndt S, Hardouin J, Garand M, Ardidie‐Robouant C, Barret A, Melino G, Lortat‐Jacob H, Muller L, Monnot C, Germain S. The interaction of heparan sulfate proteoglycans with endothelial transglutaminase‐2 limits VEGF165‐induced angiogenesis. Sci Signal. 2015; 8:ra70.CrossrefMedlineGoogle Scholar
- Holfeld J, Tepeköylü C, Kozaryn R, Mathes W, Grimm M, Paulus P. Shock wave application to cell cultures. J Vis Exp. 2014; 86:e51076.Google Scholar
- Theurl M, Schgoer W, Albrecht K, Jeschke J, Egger M, Beer AG, Vasiljevic D, Rong S, Wolf AM, Bahlmann FH, Patsch JR, Wolf D, Schratzberger P, Mahata SK, Kirchmair R. The neuropeptide catestatin acts as a novel angiogenic cytokine via a basic fibroblast growth factor‐dependent mechanism. Circ Res. 2010; 107:1326–1335.LinkGoogle Scholar
- Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, Kass DA. Measurement of cardiac function using pressure‐volume conductance catheter technique in mice and rats. Nat Protoc. 2008; 3:1422–1434.CrossrefMedlineGoogle Scholar
- Primessnig U, Schonleitner P, Holl A, Pfeiffer S, Bracic T, Rau T, Kapl M, Stojakovic T, Glasnov T, Leineweber K, Wakula P, Antoons G, Pieske B, Heinzel FR. Novel pathomechanisms of cardiomyocyte dysfunction in a model of heart failure with preserved ejection fraction. Eur J Heart Fail. 2016; 18:987–997.CrossrefMedlineGoogle Scholar
- Siow RC. Culture of human endothelial cells from umbilical veins. Methods Mol Biol. 2012; 806:265–274.CrossrefMedlineGoogle Scholar
- Baker M, Robinson SD, Lechertier T, Barber PR, Tavora B, D’Amico G, Jones DT, Vojnovic B, Hodivala‐Dilke K. Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc. 2012; 7:89–104.CrossrefGoogle Scholar
- Albrecht‐Schgoer K, Schgoer W, Holfeld J, Theurl M, Wiedemann D, Steger C, Gupta R, Semsroth S, Fischer‐Colbrie R, Beer AG, Stanzl U, Huber E, Misener S, Dejaco D, Kishore R, Pachinger O, Grimm M, Bonaros N, Kirchmair R. The angiogenic factor secretoneurin induces coronary angiogenesis in a model of myocardial infarction by stimulation of vascular endothelial growth factor signaling in endothelial cells. Circulation. 2012; 126:2491–2501.LinkGoogle Scholar
- Holfeld J, Tepekoylu C, Reissig C, Lobenwein D, Scheller B, Kirchmair E, Kozaryn R, Albrecht‐Schgoer K, Krapf C, Zins K, Urbschat A, Zacharowski K, Grimm M, Kirchmair R, Paulus P. Toll‐like receptor 3 signalling mediates angiogenic response upon shock wave treatment of ischaemic muscle. Cardiovasc Res. 2016; 109:331–343.CrossrefMedlineGoogle Scholar
- Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LEJ, Berman D, Czer LSC, Marbán L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marbán E. Intracoronary cardiosphere‐derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012; 379:895–904.CrossrefMedlineGoogle Scholar
- Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 1992; 267:26031–26037.CrossrefMedlineGoogle Scholar
- Uwatoku T, Ito K, Abe K, Oi K, Hizume T, Sunagawa K, Shimokawa H. Extracorporeal cardiac shock wave therapy improves left ventricular remodeling after acute myocardial infarction in pigs. Coron Artery Dis. 2007; 18:397–404.CrossrefMedlineGoogle Scholar
- Hatanaka K, Ito K, Shindo T, Kagaya Y, Ogata T, Eguchi K, Kurosawa R, Shimokawa H. Molecular mechanisms of the angiogenic effects of low‐energy shock wave therapy: roles of mechanotransduction. Am J Physiol Cell Physiol. 2016; 311:C378–C385.CrossrefMedlineGoogle Scholar
- Burneikaite G, Shkolnik E, Celutkiene J, Zuoziene G, Butkuviene I, Petrauskiene B, Serpytis P, Laucevicius A, Lerman A. Cardiac shock‐wave therapy in the treatment of coronary artery disease: systematic review and meta‐analysis. Cardiovasc Ultrasound. 2017; 15:11.CrossrefMedlineGoogle Scholar
Can Gollmann‐Tepeköylü, Daniela Lobenwein, Markus Theurl, Uwe Primessnig, Daniela Lener, Elke Kirchmair, Wolfgang Mathes, Michael Graber, Leo Pölzl, Angela An, Katarzyna Koziel, Elisabeth Pechriggl, Jakob Voelkl, Patrick Paulus, Wolfgang Schaden, Michael Grimm, Rudolf Kirchmair and Johannes Holfeld See fewer authors